Partial Central Adrenal Insufficiency Due to Possibly Autoimmune Hypophysitis, Misdiagnosed as Depression
*Corresponding Author(s):
Pamela R Chavez-DiazDepartment Of Endocrinology, Hospital Universitario La Paz, Paseo De La Castellana, Madrid, Spain
Tel:+34 911285223,
Email:rossch23@gmail.com
Miriam Zapatero-Larrauri
Department Of Endocrinology, Hospital Universitario La Paz, Paseo De La Castellana, Madrid, Spain
Tel:+34 911285223,
Email:miriamzlarrauri@gmail.com
Abstract
A 22 year-old Spanish female with type 1 diabetes, who had been diagnosed with severe anxious depressive syndrome, was treated with anti-depressants and antipsychotics for the previous four years with no improvement of her general condition. Endocrinology examination revealed incongruous low-normal levels of plasma ACTH with low cortisol. Intravenous ACTH 250 ug stimulation test for cortisol and 17-OH progesterone was normal. Pituitary MRI displayed that stalk was slightly pulled to the left and had a slight thickening in the upper third. With great awareness of partial failure adrenal, we started steroids. Few weeks later psychiatric symptoms improved. Two years afterwards, the patient developed autoimmune subclinical hypothyroidism. Two previous immune aggregation guided us to another autoimmune process. In conclusion, adrenal insufficiency may be misdiagnosed as depression. It is important to consider this diagnosis in similar cases.
Keywords
INTRODUCTION
Hypophysitis is a rare disease that consists of lymphocytic infiltration of the stalk and pituitary gland. It has been classified according to the anatomic location of the infiltrate (adenohypophysitis, infundibuloneurohypophysitis or panhypophysitis), histopathology (lymphocytic, granulomatous and rarely xanthomatous, Ig G4 related necrotizing or mixed forms) and etiology (primary or secondary to sellar diseases, systemic diseases as sarcoidosis, langerhans cell histiocytosis, tuberculosis, syphilis or drugs). It has been associated with other autoimmune diseases such as hashimoto thyroiditis, autoimmune polyglandular syndrome, Graves’ disease, systemic lupus erythematosus, Sjögren syndrome, Wegener’s granulomatosis and type 1 diabetes. New categories have recently been established such as Ig G4 related hypophysitis and secondary hypophysitis by anticytotoxic T lymphocyte antigen 4 antibodies [1-5].
The prevalence and incidence are probably underestimated. The literature reports a prevalence of 0.24 to 0.88% and annual incidence around 0.1 per million. The most common form of hypophysitis is the lymphocytic one (71.8%). The mean age at the onset is around 40 years old. Lymphocytic adenohypophysitis is more common in women especially during peripartum period.
Peripartum period and aseptic meningitis have been established as risk factors for primary hypophysitis [6]. The underlying pathophysiology has an autoimmunity basis in the peripartum period, since hyperestrogenism produces an increased volume and perfusion of the gland, exposing it to the immune system [2]. The finding of antipituitary antibodies, albeit with low sensitivity and specificity (26 and 36% respectively) [1-7], supports this theory.
The natural course of the disease develops in phases. Initially, inflammation produces only a subclinical mass effect. Then, depending on whether the inflammation subsides or progresses?the patient may develop hypopituitarism, which is the most frequent consequence. The sellar mass effect induces headache, visual impairment and diplopia. The most common hormonal deficiencies affect ACTH, TSH and rarely LH, FSH, prolactin, and GH secretion. Central diabetes insipidus occurs mainly in infundibuloneurohypophysitis and panhypophysitis. Besides hyperprolactinemia can be found due to stalk compression, direct damage or decreased hypothalamic dopamine secretion.
As mentioned above, antipituitary antibodies are not a valid diagnostic test for hypophysitis and might be present in other autoimmune diseases such as Hashimoto’s thyroiditis, Graves’ disease, type 1 diabetes or Cushing’s disease. Magnetic Resonance Image (MRI) is characterized by symmetric enlargement of the pituitary stalk or pituitary gland, with a strong homogeneous gadolinium enhancement. The definitive diagnosis would be given by histopathology if available [8].
The clinical diagnosis is based on three or more of the following criteria [2]: 1) women at peripartum period, 2) age below 30 years old, 3) hormonal pituitary deficiency, 4) presence of other autoimmune diseases or autoimmune markers, 5) acute onset of headache or other mass effect symptoms, 6) acute diabetes insipidus, 7) positive antipituitary antibodies, 8) linfomonocytic pleocytosis in the cerebrospinal fluid, 9) characteristic MRI.
The treatment has two objectives: treatment of mass effect and hormone replacement. In the presence of compressive symptoms (optical nerve compression or intracranial hypertension) transphenoidal surgery should be considered [2,3]. Otherwise, treatment with corticosteroids may decrease the enlargement of the gland. Stereotactic radiosurgery is another option in selected cases. The prognosis is variable depending on evolution time, residual fibrosis and response to treatment.
CASE REPORT
A 22 year-old Spanish female was diagnosed with type 1 diabetes when she was fourteen, based on hyperglycemia, positive anti-pancreatic islet antibodies (Anti- GAD65, Anti-IA2) and an initial C peptide of 0,7 ng/mL (0.5-3.5) maintaining a good metabolic control (HbA1C less than 6.5%). At the age of 15, she started with asthenia and depressive symptoms, being diagnosed as having a severe anxious depressive syndrome three years later. Since then, antidepressants, anxiolytic and antipsychotics, such as sertraline, aripiprazole and bromazepam, were prescribed. However, her general condition did not improve. On the other hand, she denied polydipsia, polyuria or nocturia. She did not have medical history of tuberculosis or another granulomatous disease, dacryoadenitis or xerostomy and steroids had never taken.
Physical examination revealed a normal blood pressure (107/60 mmHg), 53 kg of weight and a BMI of 22 kg/m2 with adequate pubertal development and normal menses. She did not present goitre, hyper pigmentation, hirsutism or acne.
Biochemical analysis showed normal levels of sodium and potassium. Endocrinological examinations showed plasma ACTH and basal cortisol levels of 8 pg/mL (normal range: 5-46 pg/mL) and 4.3 ug/dL (normal range: 5, 0-25 ug/dL), respectively, with similar levels in three determinations. It was striking as ACTH remained at the low normal range despite low cortisol levels.
An intravenous 250 ug-ACTH stimulation test for cortisol and 17-OH progesterone produced a normal response with initial cortisol of 4.3 ug/dL, 30 min after ACTH of 19.9 ug/dL and 60 min after of 22.6 ug/dL (5.0-25). Levels of free thyroxin and TSH, gonadotropins, prolactin, GH and IGF-1 were normal. Antiperoxidase and thyroglobulin antibodies were positive. The antipituitary antibodies determination was negative.
The T1-weighted magnetic resonance imaging of the pituitary gland showed that pituitary stalk was slightly pulled to the left and had a slight thickening in the upper third (Figure 1).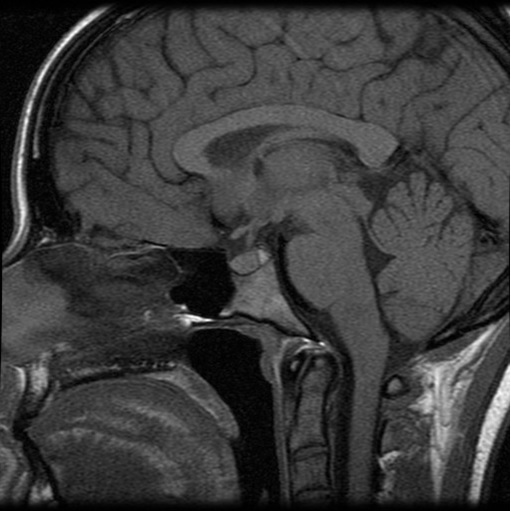
Figure 1: This is the sagittal magnetic resonance image of the patient showing a slight thickening in the upper third of stalk pituitary (white arrow).
The suspicion of a partial failure ACTH was high, so we decided starting glucocorticoid treatment (5 mg/day of hydrocortisone). After few weeks, the patient experienced a great symptomatic improvement, keeping only low doses of sertraline. Last control after a 24-hour discontinuation of hydrocortisone showed a 2.9 μg/dL basal cortisol at 9 am (normal range: 5.0-25 ug/dL) that confirmed the diagnosis.
Two years later levothyroxine was prescribed because of subclinical hypothyroidism. Hydrocortisone requirement remained stable after levothyroxine treatment.
DISCUSSION
Due to the fact that depression symptoms are usually nonspecific; a wide differential diagnosis is needed. In our case, the patient remained paucisymptomatic, only showing depressive symptoms [4]. Bad response to antidepressants should alert us of a possible underlying pathology, considering adrenal insufficiency among others.
The Incongruous low-normal ACTH for low cortisol as three times alerted us about a central failure of adrenal axis. On the other hand, normal response to exogenous ACTH allowed us to maintain this suspicion, it was practically confirmed by the improvement of symptoms with hydrocortisone treatment plus the hypocortisolism relapse after hydrocortisone withdrawal and MRI findings. We use the term “partial” as due to normal ACTH levels.
Likewise, despite of the nonspecific symptoms, the presence of two autoimmune disease as type 1 diabetes and tyroiditis gave us the clue to consider an additional autoimmune disorder located in adrenal axis. Another option we should consider was a IgG4-related hypophysitis. IgG4-related disease is characterized by marked infiltration of lymphocytes and fibrosis in several organs, including pancreas, hypophysis, lacrimal and salivary glands, thyroid, liver, kidney and lung [9]. In our case, the absence of these clinical manifestations made less likely this entity, although we could not dismiss it due to the unavailabilty of laboratory markers such as IgG4.
In this patient we excluded polyglandular syndrome due to the fact that the suprarrenal failure was central instead of primary. Besides, rarely hypopituitarism has been described as part of this syndrome.
In our case the autoimmune thyroiditis developed after the pituitary disease. It is described that these two conditions may coexist or thyroid disease may precede or be diagnosed after [2]. Moreover, the mild radiologic findings supported this conclusion, despite the absence of antipituitaty antibodies, although other etiologies cannot be ruled without performing a biopsy.
CONCLUSION
Adrenal insufficiency is not unusual, may be paucisymptomatic and could be misdiagnosed with depression. We suggest considering this diagnosis in young women with depressive symptoms and an underlying autoimmune disease.
REFERENCES
- Fukuoka H (2015) Hypophysitis. Endocrinol Metab Clin North Am 44: 143-149.
- Català Bauset M, Gilsanz Peral A, Girbés Borràs J, Zugasti Murillo A, Moreno Esteban B, et al. (2008) Clinical practice guideline for the diagnosis and treatment of hypophysitis. Endocrinol Nutr 55: 44- 53.
- Laws ER, Vance ML, Jane JA Jr (2006) Hypophysitis. Pituitary 9: 331-333.
- Hiroi N, Yoshihara A, Sue M, Yoshino G, Higa M (2010) Central Adrenal Insufficiency and Diabetes Insipidus Misdiagnosed as Severe Depression. Clin Med Insights Case Rep 3: 55-58.
- Leporati P, Landek-Salgado MA, Lupi I, Chiovato L, Caturegli P (2011) IgG4-related hypophysitis: a new addition to the hypophysitis spectrum. J Clin Endocrinol Metab 96: 1971-1980.
- Suzuki K, Izawa N, Nakamura T, Hashimoto K, Ouchi K, et al. (2011) Lymphocytic hypophysitis accompanied by aseptic meningitis mimics subacute meningoencephalitis. Intern Med 50: 2025-2030.
- De Bellis A, Pane E, Bellastella G, Sinisi AA, Colella C, et al. (2011) Detection of antipituitary and antihypothalamus antibodies to investigate the role of pituitary or hypothalamic autoimmunity in patients with selective idiopathic hypopituitarism. Clin Endocrinol (Oxf) 75: 361-366.
- Matoba K, Mitsuishi S, Hayashida S, Yamazaki H (2014) Hypopituitarism possibly due to lymphocytic hypophysitis in a patient with type 1 diabetes. Intern Med 53: 1961-1964.
- Umehara H, Nakajima A, Nakamura T, Kawanami T, Tanaka M, et al. (2014) IgG4-related disease and its pathogenesis-cross-talk between innate and acquired immunity. Int Immunol 26: 585-595.
Citation: Chávez-Díaz PR, Zapatero-Larrauri M, Barquiel B, Utrilla C, Doforno R, et al. (2016) Partial Central Adrenal Insufficiency Due to Possibly Autoimmune Hypophysitis, Misdiagnosed as Depression. J Clin Stud Med Case Rep 3: 030.
Copyright: © 2016 Pamela R Chavez-Diaz, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.