Preparing and Processing Complex Tissue Constructs Composed of Cellular Spheroids for Histological Examination
*Corresponding Author(s):
Alexis FDepartment Of Bioengineering, Clemson University, 301 Rhodes Research Center, Clemson, South Carolina, United States
Tel:8646565003,
Fax:8646564466
Email:falexis@clemson.edu
Abstract
Using cellular spheroids to build tissues and organs is a rapidly emerging technology that will have great impact on the biomedical industry. As tissues are fabricated in vitro, it will be critical to histologically examine tissue composition, function and organization for comparison with native tissues. Conventional automated histological methods are not recommended for fixation and processing of tissues composed of spheroids due to their delicate nature and the need to preserve tissue morphology. Given this, there is a need to develop a manual method to prepare and process tissues composed of cellular spheroids for histological sectioning and examination. Here, we report a manual processing technique developed for preparing larger tissues composed of spheroids for histological examination based on our lab's work using magnetic forces to pattern and assemble tissues composed of Janus Magnetic Cellular Spheroids for tissue engineering applications. The results presented here contribute to understanding the organization of cells, studying extracellular matrix content, analyzing phenotypic expression and localizing iron oxide magnetic nanoparticles in tissues composed of Janus Magnetic Cellular Spheroids.
Keywords
INTRODUCTION
Tissue engineering uses knowledge of biology, material science, biochemistry, engineering and physiological systems to replace or repair damaged, diseased or lost tissues in the human body with functional tissue that mimics native tissue. One of the biggest challenges in the field of tissue engineering is the in vitro fabrication of functional tissue engineered constructs with high cell density, high cell viability and an Extracellular Matrix (ECM) network that mimics native tissue. Three dimensional cell cultures, or spheroids, are being investigated as building blocks for scaffold-free tissue engineering applications due to their ability to mimic the native cellular and ECM environment of natural tissues [1]. Spheroids can be fabricated with one cell type, or co-cultured with other cell types, which is desirable for fabricating tissue types with varying complexities and functionalities [2,3]. Tissue assembly and fabrication methods include 3D printing, cell sheet techniques, and patterned molds [4-9]. These methods assemble the spheroids or cells into a desired position through passive contact, but do not encourage active contact mediated by forces to promote fusion of the tissue over time. This can cause issues with tissue homogeneity, can alter tissue geometry and can lead to increases in fabrication time. Our lab has developed the Janus Magnetic Cellular Spheroid (JMCS) structure to safely incorporate iron oxide Magnetic Nanoparticles (MNPs) into cellular spheroids [10-13]. This method allows for spatial control over MNPs to form two distinct domains within the spheroid: (1) cells and ECM and (2) extracellular MNPs. Safely incorporating MNPs allows for utilizing magnetic forces at a distance to pattern JMCSs into desired geometries for building larger, more complex tissue constructs such as rings, spheroid sheets and tubes for various tissue engineering applications [12].
Further, it is critical to understand and visualize the fusion and maturation of tissues composed of JMCSs over time. To understand these mechanisms, studies must include analyzing phenotypic expression, visualizing the spatial orientation and viability of cells, studying ECM organization and localizing iron oxide MNPs throughout the developing tissues over time. Tissue stains, such as the Hemotoxylin and Eosin (H&E) stain, the Masson's Trichrome stain, the Verhoeff-Van Gieson stain, Turnbull's Blue Reaction stain, and immunohistochemical stains have been developed for these purposes. While histological examination allows for analysis of the tissues composed of spheroids, preparing and processing these tissues for staining and analysis is challenging. It is not recommended to use conventional histological processors or standard processing methods, due to the delicacy of the samples, which makes it necessary to use manual processing to increase the survival of the samples and preserve tissue morphology. Through our work, we encountered the need for extensive histological analysis of assembled tissue constructs. Here, we report the techniques developed in our lab for use in the processing of larger, complex tissues for histological examination.
MATERIALS AND METHODS
Cell culture
Spheroid assembly
Fabrication of tissue rings
Fabrication of tissue sheets
Fabrication of tissue tubes
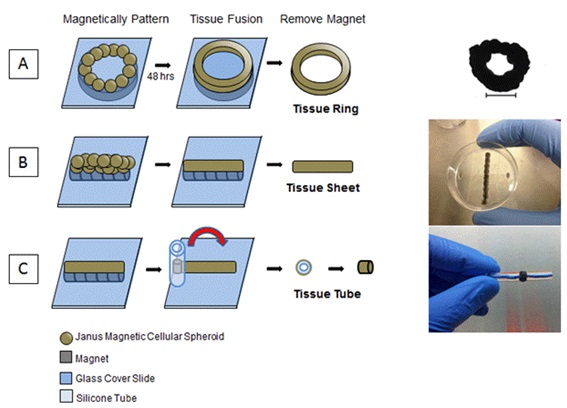 Figure 1: Magnetic force assembly of JMCSs to fabricate tissue rings, sheets and tubes.
Figure 1: Magnetic force assembly of JMCSs to fabricate tissue rings, sheets and tubes.
Tissue fixation, processing, embedding and sectioning
After overnight fixation, the Z-Fix was removed and the tissue samples were dehydrated using a series of ethanol and xylene incubations (Table 1) [14]. Immediately prior to the first ethanol incubation, the tissue tube was removed from the silicone mandrel. Given that our lab has done extensive histological examination of spheroids, a protocol was developed for preparing and processing these individual spheroids. However, since the tissue rings, tissue sheets and tissue tubes are larger, more complex and composed of many spheroids, the times for the ethanol incubations were doubled to ensure proper infiltration. For ethanol solutions, deionized and distilled water were used for dilutions. The times selected for incubation were chosen in accordance with literature values with automated and manual processing [15]. The times used for incubation are also comparable to what automated tissue processing programs regularly use (SAKURA Tissue TEK VIP) for standard tissue processing. After incubation in xylene, the solution was removed and replaced with molten paraffin (Surgipath EM-400, 56°C, Leica, Illinois, USA) using a Tissue-Tek TEC Tissue Embedding Console System (Sakura, California, USA). Samples submerged in molten paraffin were placed in one of the thermal chambers of the embedding console for overnight infiltration of paraffin at 60°C.
| Step | Reagents | Time |
| 1 | Z-fix | Overnight |
| 2 | 70% Ethanol | 30 minutes |
| 3 | 80% Ethanol | 30 minutes |
| 4 | 95% Ethanol | 30 minutes |
| 5 | 95% Ethanol | 30 minutes |
| 6 | 100% Ethanol | 30 minutes |
| 7 | 100% Ethanol | 30 minutes |
| 8 | Xylene | 15 minutes |
| 9 | Moltene Paraffin | Overnight |
After overnight incubation in molten paraffin, tissue samples were transferred to appropriately sizedem bedding molds filled with molten paraffin. Here, care was taken to properly orient the tissues in the center of the mold for ease of sectioning. The embedding molds were each covered with a numbered embedding block holder and placed on a cold plate at -4°C for at least 1 hour. Following, the molds were removed and embedded tissue blocks were placed in an ice water bath for 10 minutes in order to rehydrate the tissue samples for ease of sectioning. A microtome was used to cut 5 µm thin sections from the tissue samples. These sections were suspended on the surface of a heated deionized water bath (44°C) and placed on a silanized glass histology slides (Adhesive Coated Slides, New Silane White Frosted, Newcomer Supply, Wisconsin, USA), due to the delicacy of the tissue samples [16]. Each slide was visually examined under a microscope to see if the sample was observed and to check for tears, folds or irregularities of the sample. The slides containing samples were incubated overnight in an oven (65°C) to remove excess paraffin.
Hematoxylin and eosin stain
Masson's trichrome stain
Tissue sheet viability
Immunohistochemistry for functional protein markers
Imaging
RESULTS
Histological examination of the tissue sheets and tubes shows that the manual processing method yielded intact tissue structures with preserved morphology. Tissue sheet sections were stained using an H&E stain and demonstrate the presence of cell nuclei throughout the fused tissues after 3 days (Figure 2A). The Masson's Trichrome stain demonstrates the presence of collagenous ECM throughout the fused tissue (Figure 2B). A LIVE/DEAD stain of the fused tissue sheet qualitatively confirms the presence of viable cells throughout the tissue, as seen by the large population of viable (green) cells with only a small portion of dead (red) cells (Figure 2C). Tissue tube sections were also stained using H&E stain and demonstrate the presence of cell nuclei throughout the fused tissue after 7 days (Figure 3A). The Masson's Trichrome stain demonstrates that collagenous ECM is present throughout the tissue tube (Figure 3B). Tissue tubes were fluorescently stained for known phenotypic smooth muscle cell markers: Smooth Muscle Myosin Heavy Chain (SMHC) (Figure 3C), Smooth Muscle 22 (SM22) (Figure 3D) and Smooth Muscle Alpha Actin (SMAA) (Figure 3E). Results demonstrate positive expression of each of these key phenotypic markers in the tissue tubes (red for SMHC and SM22, green for SMAA), as well as the presence of nuclei throughout the tissue using a Hoechst stain for nuclei (blue).
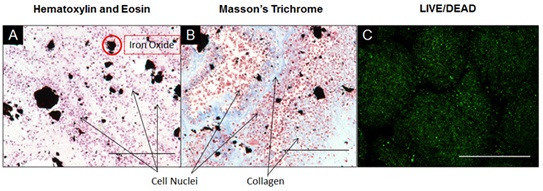 Figure 2: Histological Examination of Tissue Sheets Composed of JMCSs.
Figure 2: Histological Examination of Tissue Sheets Composed of JMCSs.
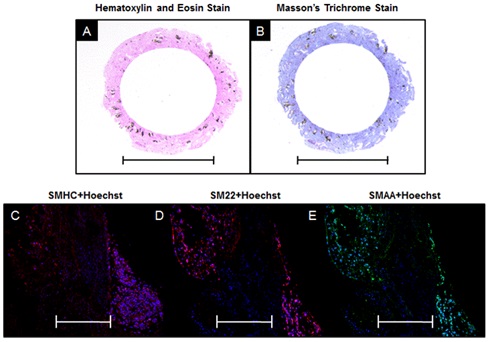 Figure 3: Histological Examination of Tissue Tubes Composed of JMCSs.
Figure 3: Histological Examination of Tissue Tubes Composed of JMCSs.DISCUSSION
Cellular spheroids are attractive for use as the building blocks for larger and more complex tissues because they mimic the 3D native orientation of tissues and produce their own ECM over time [7,17]. The bottom up, scaffold free tissue engineering approach with cellular spheroids is promising for the fabrication of larger tissues because industrial systems are being developed to robotically scale tissue and organ fabrication using automated spheroid bioprinters [17]. To compare tissues fabricated in vitro from spheroids to native tissues, it is critical to develop an automated method to rapidly process these tissues for histological examination of tissue structure, cellular orientation and ECM organization. Tissues composed of spheroids are subject to poor mechanical properties, due to their fluidic nature and lack of maturation [18]. Thus, care must be taken with these delicate samples to preserve tissue integrity and tissue morphology during the processing steps.
In this work, a manual histological processing technique for tissues composed of spheroids was presented. This technique uses overnight fixation and also overnight infiltration in paraffin. The chosen manual method of processing has been compared to automatic processing with a SAKURA Tissue TEK VIP. When using the automatic processor, spheroid samples have been lost and tissues composed of spheroids have had their morphologies compromised. This is especially critical for tissue tubes composed of spheroids, where it is important to maintain the morphology of the lumen for further analysis of cells, markers, and ECM distribution. In general, tissue samples composed of spheroids are delicate and the manual processing allows for proper care to be taken. Manual processing allows for careful solution exchanges and prevents samples from being lost because the sample can always been visualized. This research offers a reliable method for preparing tissues to study the localization and viability of cells, the organization of extracellular matrix and phenotypic expression of tissues composed of cellular spheroids. This understanding is critical when developing tissue engineered constructs that mimic native tissue. Though the method is sufficient for small scale histological analyses, an automated histological processer must be developed in the future to meet the need at the industrial scale. Morales et al., developed a method to rapidly fix, process and embed tissue specimens in 1 hour by using a continuous flow of the working solutions [19]. Another strategy for making the process more high-throughput could be to embed multiple spheroid samples throughout the same paraffin block, which reduces the amount of blocks to be made and sectioned [20].
Using the manual technique presented here, tissues of various shape, size and composition were able to be processed and prepared for histological examination. Sectioned specimens were stained using H&E, Masson's Trichrome, LIVE/DEAD and various IHC reagents. The H&E stain allowed for visualization of cell nuclei, the Masson's Trichrome stain highlighted collagen throughout the tissue, the LIVE/DEAD stain demonstrated high cell viability in tissue sheets and IHC staining demonstrated phenotypic expression expected of smooth muscle cells. These results qualify the presented manual method of tissue processing for preparing tissues for standard stains, like H&E, as well as specialized stains, like fluorescent IHC staining. The presented method allowed for visualization of cells, characterization of collagen, localization of iron oxide MNPs and analysis of phenotypic expression throughout tissues composed of cellular spheroids. Having a full understanding of these tissue molecules and components is critical for developing a tissue engineered construct that mimics native tissue.
ACKNOWLEDGEMENTS
This work was supported by the American Heart Association Beginning Grant in Aid-2BGIA11720004 award to F.A. and the SC EPSCOR Grant for Exploratory Academic Research-2012001188 awarded to F.A. The authors wish to thank Dr. T. Bruce and the Clemson Light Imaging Facility at Clemson University for technical support with microscopy.
REFERENCES
- Olsen TR, Frank A (2014) Bioprocessing of Tissues Using Cellular Spheroids. J Bioproces Biotechniq 4: 112.
- Mironov V, Prestwich G, Forgacs G (2007) Bioprinting living structures. J Mater Chem 17: 2054-2060.
- Jakab K, Neagu A, Mironov V, Markwald RR, Forgacs G (2004) Engineering biological structures of prescribed shape using self-assembling multicellular systems. Proc Natl Acad Sci USA 101: 2864-2869.
- Boland T, Xu T, Damon B, Cui X (2006) Application of inkjet printing to tissue engineering. Biotechnol J 1: 910-917.
- L'heureux N, Pâquet S, Labbé R, Germain L, Auger FA (1998) A completely biological tissue-engineered human blood vessel. FASEB J 12: 47-56.
- Jakab K, Norotte C, Marga F, Murphy K, Vunjak-Novakovic G, et al., (2010) Tissue engineering by self-assembly and bio-printing of living cells. Biofabrication 2: 022001.
- Mironov V, Visconti RP, Kasyanov V, Forgacs G, Drake CJ, et al., (2009) Organ printing: Tissue spheroids as building blocks. Biomaterials 30: 2164-2174.
- Tan Y, Richards DJ, Trusk TC, Visconti RP, Yost MJ, et al., (2014) 3D printing facilitated scaffold-free tissue unit fabrication. Biofabrication 6: 024111.
- Jakab K, Damon B, Neagu A, Kachurin A, Forgacs G (2006) Three-dimensional tissue constructs built by bioprinting. Biorheology 43: 509-513.
- Mattix B, Olsen TR, Moore T, Casco M, Simionescu D, et al., (2014) Accelerated Iron Oxide Nanoparticle Degradation Mediated by Polyester Encapsulation within Cellular Spheroids. Adv Funct Mater 24: 800-807.
- Mattix B, Olsen TR, Gu Y, Casco M, Herbst A, et al., (2014) Biological magnetic cellular spheroids as building blocks for tissue engineering. Acta Biomater 10: 623-629.
- Mattix BM, Olsen TR, Casco M, Reese L, Poole JT, et al., (2014) Janus magnetic cellular spheroids for vascular tissue engineering. Biomaterials 35: 949-960.
- Mattix B, Poole T, Casco M, Uvarov O, Visconti, RP, et al., (2013) Small Diameter Vascular Grafts: Replacement Strategies, Maturation Techniques and Challenges. Journal of Tissue Science and Engineering 4: 1.
- Olsen TR, Mattix B, Casco M, Herbst A, Williams C, et al., (2014) Processing cellular spheroids for histological examination. Journal of Histotechnology.
- Buesa RJ, Peshkov MV (2009) Histology without xylene. Ann Diagn Pathol 13: 246-256.
- Lima FdO, Almeida JSd, Costa HdO, Pinheiro NF, Oshima CT, et al., (2011) Loss of samples in the tissue microarray technique: comparison between slides using adhesive tape and silanized slides. Journal of Histotechnology 34: 69-73.
- Mironov V, Kasyanov V, Markwald RR (2011) Organ printing: from bioprinter to organ biofabrication line. Curr Opin Biotechnol 22: 667-673.
- Hajdu Z, Mironov V, Mehesz AN, Norris RA, Markwald RR, et al., (2010) Tissue spheroid fusion-based in vitro screening assays for analysis of tissue maturation. J tissue eng regen med 4: 659-664.
- Morales AR, Essenfeld H, Ervin E, Duboue MC, Vincek V, et al., (2002) Continuous-specimen-flow, high-throughput, 1-hour tissue processing: System for rapid diagnostic tissue preparation. Arch Pathol Lab Med 126: 583-590.
- Jensen T, Cessna M, Miller D, Hansen J (2014) Tissue microarray advanced techniques for sampling donor blocks with limited tissues - introduction to 'the radical-TMA'. Journal of Histotechnology 37: 9-13.
Citation: Olsen TR, Casco M, Mattix B, Williams C, Herbst A, et al. (2014) Preparing and Processing Complex Tissue Constructs Composed of Cellular Spheroids for Histological Examination. J Cytol Tissue Biol 1: 002.
Copyright: © 2014 Olsen TR, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.