Progressive Microcephaly, Spasticity and Development Delay: Novel SLC1A4 Variants in Two Portuguese Families and Literature Review
*Corresponding Author(s):
Joana TeixeiraFaculty Of Medicine, University Of Porto, Portugal
Tel:351 914613157,
Email:joanafteixeira@live.com.pt
Abstract
The role of SLC1A4 gene on the L-serine transport from astrocytes to neurons (via Na+ dependent transporter system ASC) is already an established physiological mechanism. Disease-causing variants in the SLC1A4 gene have been identified as a very rare cause of neurodevelopmental disorder (spastic tetraplegia, thin corpus callosum, and progressive microcephaly - SPATCCM, OMIM 616657), mostly reported in patients with an Ashkenazi-Jewish background (homozygous for the p.(Glu256Lys) variant) (n=15 of the 17 described cases).
We hereby report two Portuguese cases presenting with significant global developmental delay, severe progressive microcephaly, spasticity and no Ashkenazi-Jewish ancestry.
Whole exome sequencing identified three novel SLC1A4 variants as the likely cause of the disease. Patient 1 is heterozygous for two splice-site pathogenic variants, c.527+1G>T and c.1035-1G>T. The impact on splicing was confirmed by a minigene strategy. Patient 2 is homozygous for a probably deleterious missense variant, c.272T>C (p.(Leu91Pro)), classified as of uncertain clinical significance.
Our results support the deleterious role of SLC1A4 variants and highlight the need of considering this entity, regardless of ethnicity, in pediatric patients presenting with unexplained neurodevelopmental delay and progressive microcephaly, associated or not with spastic tetraplegia or early onset epileptic encephalopathy.
Keywords
Developmental delay; Microcephaly; SLC1A4 gene; Whole-exome sequencing
INTRODUCTION
Neurodevelopmental and neurometabolic childhood disorders present a wide spectrum of non-specific early onset abnormalities, including, but not confined to, intellectual disability, microcephaly, spasticity, epilepsy and brain malformations.
Next-generation sequencing technologies have paved the way to uncover a growing number of etiologies and the subsequent report of new syndromes.
L-serine is essential for normal neurodevelopment and its metabolism is highly dependent on the Na+ dependent transporter system ASC (ASCT1). Deleterious variants of the transporter-encoding SLC1A4 gene are extremely rare but have been found to cause severe neurodevelopmental disorders without specific symptom patterns [1].
To date, this genetic condition has been reported almost exclusively in a limited number of Ashkenazi-Jewish (AJ) individuals and almost all homozygous for the p(Glu256Lys) variant. Until now, 19 patients with SLC1A4 disease-causing variants were described, and only four of them are not Ashkenazi-Jewish (2 Pakistani, 1 Irish and 1 Italian) (Table 1).
|
Patient/Variant Reports
|
Damseh et al
|
Damseh et al |
Damseh et al (9 patients) |
Srour et al
|
Srour et al
|
Heimer et al
|
Heimer et al
|
Conroy et al
|
Pironti et al
|
Abdelrahman et al
|
Abdelrahman et al
|
Current Patient 1
|
Current Patient 2
|
|
|
|||||||||||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Age |
3.5 years |
4.5 years |
3-15 years |
11 years |
4 years |
6 years |
4.5 years |
3 years |
7 years |
7 years |
3 years |
9 years |
9 years |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Gender |
Female |
Female |
4 males/5 |
Female |
Male |
Female |
Female |
Male |
Male |
Male |
Female |
Male |
Female |
|
|
|
|
females |
|
|
|
|
|
|
|
|
|
|
|
Ethnic group |
AJ |
AJ |
AJ |
AJ |
AJ |
AJ |
AJ-Iraqi |
Irish |
Italian |
Pakistani |
Pakistani |
Portuguese |
Portuguese |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Pregnancy |
NA |
NA |
NA |
- |
- |
- |
- |
NA |
- |
oligohydramnios |
- |
- |
- |
|
events |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Dysmorphic |
- |
- |
- |
- |
- |
- |
- |
NA |
Large nose |
Hypertelorism, |
Hypertelorism, |
Broad flat philtrum, |
- |
|
feautures |
|
|
|
|
|
|
|
|
root, low |
synophrys, |
synophrys, |
largemouth, thin |
|
|
|
|
|
|
|
|
|
|
|
implanted |
depressed nasal |
depressed nasal |
upper lip and |
|
|
|
|
|
|
|
|
|
|
|
and wide |
bridge, and |
bridge, and |
everted lower lip |
|
|
|
|
|
|
|
|
|
|
|
auricles |
prominent ears |
prominent ears |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Microcephaly |
Acquired |
Primary |
4 primary, 4 |
Acquired |
Acquired |
Acquired |
Acquired |
Primary |
Primary |
Primary |
Primary |
Acquired |
Acquired |
|
|
|
|
acquired, 1 NA |
|
|
|
|
|
|
|
|
|
|
|
Age of epilepsy |
- |
NA |
NA |
- |
- |
1 year |
11 months |
5 months |
4 months |
6 months |
9 months |
- |
5 years |
|
onset |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Type of seizure |
- |
Infantile |
2 starring |
- |
- |
NA |
Myoclonic |
Focal |
Tonic |
Generalized tonic- |
Generalized tonic- |
- |
Myoclonic |
|
|
|
spasms |
episodes, 1 |
|
|
|
|
motor and |
extensor |
clonic seizures with |
clonic seizures |
|
|
|
|
|
|
|
|
|
spasms and |
eye uprolling and |
with eye uprolling |
|
|
|||
|
|
|
|
infantile |
|
|
|
|
dyscognitiv |
|
|
|||
|
|
|
|
|
|
|
|
left eye |
loss of |
and loss of |
|
|
||
|
|
|
|
spasms, rest no |
|
|
|
|
e seizures |
|
|
|||
|
|
|
|
|
|
|
|
derivation |
consciousness |
consciousness |
|
|
||
|
|
|
|
seizures |
|
|
|
|
|
|
|
|||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Deep reflexes |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
Increased |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Tone |
Hypotonic |
Hypotonic |
5 hypotonic, 4 |
Peripheral |
Mild |
Peripheral |
Hypertonic |
Hypotonic |
Central |
Hypertonic |
Central hypotonia, |
Peripheral |
Normal tone |
|
|
|
|
hypertonic, 1 |
hypertonia |
hypertonia |
hypertonia |
|
|
hypotonia, |
|
peripheral |
hypertonia |
|
|
|
|
|
|
|
peripheral |
|
hypertonia |
|
|
||||
|
|
|
|
normotonic |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
hypertonia |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Clonus |
- |
- |
- |
+ |
- |
- |
- |
- |
NA |
+ |
+ |
- |
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Swallowing |
+ |
NA |
NA |
- |
- |
- |
- |
+ |
+ |
+ |
+ |
- |
- |
|
difficulties |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Bladder control |
NA |
NA |
NA |
NA |
NA |
NA |
NA |
NA |
NA |
- |
- |
- |
+ |
|
Motor delay |
None |
Can sit, not |
All delayed, |
Can crawl, |
Can stand, |
Crawls on |
Can crawl |
Can roll, no |
No head |
Some head control, |
Head control, no |
Some head |
Can walk |
|
|
|
standing |
variable |
stand with |
cannot |
hands and |
on tummy, |
sitting |
control |
no rolling, sitting or |
rolling, sitting or |
control, no rolling, |
|
|
|
|
|
standing |
standing |
crawling, sitting or |
|
|||||||
|
|
|
|
degrees |
support, |
cruise |
feet, walks |
no sitting |
|
|
|
|||
|
|
|
|
|
|
|
|
standing |
|
|||||
|
|
|
|
|
cannot walk |
|
with |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
assistance |
|
|
|
|
|
|
|
|
Speech delay |
Babbles, |
Non verbal |
5 nonverbal, 3 |
Nonverbal |
Nonverbal |
Babbles, no |
No babbling |
No |
Nonverbal |
Babble, no single |
Babble, no single |
Guttural sounds |
Short phrases, |
|
|
no words |
|
babble, 2 speak |
|
|
words |
|
babbling |
|
word |
word |
and some |
attends school |
|
|
|
|
|
|
|
|
|
monosyllabic |
with an |
||||
|
|
|
|
few word |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
words |
adapted |
|
|
|
|
|
phrases |
|
|
|
|
|
|
|
|
||
|
|
|
|
|
|
|
|
|
|
|
|
|
curriculum |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Abnormal |
- |
- |
- |
NA |
NA |
|
|
NA |
NA |
|
|
|
|
|
movements |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hair pulling |
|
|
|
|
|
+ |
+ |
|
|
+ |
+ |
- |
-- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Stereotypies |
|
|
|
|
|
+ |
+ |
|
|
+ |
+ |
- |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Irritability |
|
|
|
|
|
+ |
+ |
|
|
+ |
+ |
+ |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Hyperactivity |
|
|
|
|
|
+ |
+ |
|
|
NA |
NA |
- |
+ |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Sleep disorder |
|
|
|
|
|
+ |
+ |
|
|
NA |
NA |
+ |
- |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
MRI signs |
Brain |
Brain |
Thin CC, |
Thin CC, |
Mildly |
Mild |
Thin CC, |
Hypomyelina |
Hypoplastic |
Bilateral cerebral |
NA |
Thin CC |
- |
|
|
atrophy |
atrophy |
hypomyelination |
nonspecific |
thinned |
cerebral |
delayed |
tion, thin CC |
CC, enlarged |
atrophy, atrophied |
|
|
|
|
|
anterior |
CC |
|
|
|
||||||||
|
|
and |
|
, brain atrophy |
white matter |
CC, mild |
atrophy, |
myelination, |
|
|
|
|
||
|
|
|
|
commisure, |
|
|
|
|
||||||
|
|
hypomyelin |
|
|
abnormalities |
myelination |
thin CC |
cerebral |
|
|
|
|
|
|
|
|
|
|
|
cerebral and |
|
|
|
|
|||||
|
|
ation |
|
|
|
, cerebral |
|
atrophy |
|
|
|
|
|
|
|
|
|
|
|
|
|
brain stem |
|
|
|
|
|||
|
|
|
|
|
|
atrophy |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
atrophy |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Variant |
p.Arg457Tr |
p.Glu256Ly |
p.Glu256Lys |
p.Glu256Lys |
p.Glu256L |
p.Glu256L |
p.Glu256Ly |
p.Trp453* |
p.Gly381Arg |
p.Tyr191* |
p.Tyr191* |
|
p.Leu91Pro |
|
|
p |
s and |
|
|
ys |
ys |
s and |
|
|
|
|
c.527+1G>T intron |
|
|
|
|
p.Leu315His |
|
|
|
|
p.Leu315Hisf |
|
|
|
|
2 and c.1035- |
|
|
|
|
fs*42 |
|
|
|
|
s*42 |
|
|
|
|
1G>T intron 6 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Table 1: The clinical profile of all reported patients with SLC1A4 disease-causing variants - Adapted from [2].
We present two unrelated Portuguese patients with significant global psychomotor developmental delay, severe progressive microcephaly and spasticity, with no AJ ancestry and with variants identified by whole-exome sequencing.
MATERIALS AND METHODS
This study was approved by the ethical committee of Hospital de São João (Nº365/19). Written informed consent was obtained from the legal guardians of the participants according to Portuguese Law.
DNA was extracted from peripheral blood according to standard protocols in the Genetics Service of Faculty of Medicine, University of Porto, Portugal.
Whole exome sequencing
Exome sequencing was performed using Agilent’ SureSelect Human All Exon V6 capture kit and a HiSeq sequencer (Illumina). For data analysis, a custom validated pipeline based on the Broad Institute’s Best Practices recommendations was applied, using bwa-mem for alignment to the GRCh37 build of the human genome, GATK HaplotypeCaller for variant calling and Ensembl VEP and GEMINI for annotation of the called variants. Quality control was performed on the resulting FASTQ (FastQC), BAM (QualiMap and samtools) and VCF (bcftoos) files and aggregated on a single QC report using MultiQC. For trio analysis, all variants were analyzed based on their inheritance pattern. Virtual panels (defined as the genomics regions corresponding to the gene’s coding exons ± 20 bps) were applied for the Mendeliome and disease-specific panels. Variants were then filtered by their minor allele frequency (MAF, below 1% in population databases: NCBI’s dbSNP, 1000 Genome Project, Exome Variant Server and gnomAD) and consequence, with variants that resulted in a change at the protein level and/or were previously described in the Human Gene Mutation Database (HGMD) or NCBI’s ClinVar being prioritized. All filtered variants were further analyzed using Alamut Visual (Interactive Biosoftware) for in-silico pathogenicity predictions (including SIFT, PolyPhen2, CADD and splice site predictions), population frequency confirmation and visual verification of the called variants in the alignment. After filtering, at least the lower quality variant was confirmed by Sanger sequencing.
Minigene assay
The SLC1A4 minigene constructs were obtained by cloning exonic and intronic sequences flanking the variants of interest into the pCMVdi vector, kindly provided by Alexandra Moreira, using the Gibson assembly method [3]. Briefly, exons 1 and 2 with intronic regions (for c.527+1G>T variant) or exons 5, 6 and 7 with intronic regions (for c.1035-1G>C) were PCR amplified from patient’s genomic DNA. In addition, the sequence of intron 1 was modified by site-directed mutagenesis to generate the wild-type construct, using the QuikChange II Kit (Agilent Technologies), according to the manufacturer’s protocol. HEK293T cells (kindly provided by Elsa Logarinho, IBMC/i3S, Porto) were grown in DMEM high glucose GlutaMAX™ supplemented with 10% fetal bovine serum (FBS) and 1% antibiotic-antimycotic (Gibco, Life technologies) at 37°C in a humidified 5% CO2 atmosphere. Cells were transiently transfected with the indicated constructs using jetPRIME (Polyplus-transfection) according to the manufacturer’s protocol. RNA was extracted 48h after transfection, using NZYol (Nzytech) as per manufacturer’s recommendations, followed by purification of the RNA aqueous phase using the RNeasy mini kit (Qiagen). RNA quantification was performed on NanoDrop 2000 (Thermo Scientific). cDNA was synthesized by reverse transcription-PCR of 2 µg total RNA with oligo(dT) using SuperScript III first-strand synthesis system (Invitrogen), according to the manufacturer’s protocol. The resulting cDNA was amplified by PCR using specific primers and loaded on agarose gel for extraction with the Zymoclean Gel DNA Recovery Kit (Zymo Research). The resulting products were sequenced by Sanger sequencing using Big Dye Terminator Cycle Sequencing v1.1 (Applied Biosystems) and an ABI 3130xl Genetic Analyzer (Applied Biosystems)
RESULTS
Case 1
The first patient is a 9-year-old boy, the youngest of two children born from non-consanguineous non-Ashkenazi-Jewish parents. (Figure 1). He was born at term, after an uneventful pregnancy and delivery, with a normal head circumference (OFC=34cm). Hypotonia, global developmental delay and acquired microcephaly (OFC=37cm, -3.5 SD, below the 1st percentile) became evident at 4 months and by 12 months of age, he began to present midline stereotypies (hand-washing and hand-mouthing), similar to those observed in patients with Rett Syndrome. He never acquired gait or language.
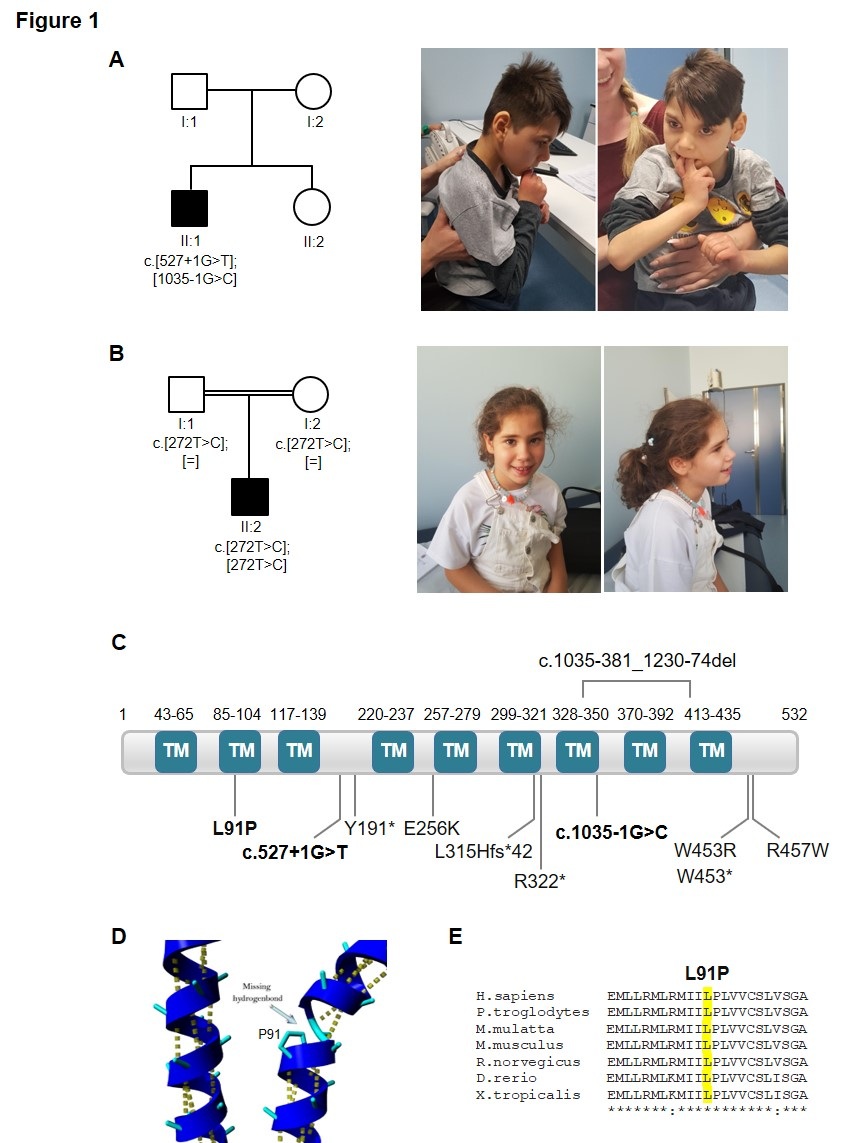
Figure 1: Families pedigrees, patient pictures and schematic representations of SLC1A4 protein. (A) Pedigree of the individual carrying the splice site variants (c.527+1G>T and c.1035-1G>C) and pictures of case 1, at 9 years of age, supported by the mother, showing microcephaly and midline stereotypies. (B) Pedigree with segregation analysis of the missense variant (c.272C>T; p.(Leu91Pro)) and pictures of case 2, at 9 years of age, sitting unsupported. Black symbols represent affected individuals. (C) Schematic representation of the SLC1A4 protein (NP_003029.2). Variants found is this study are highlighted in bold. Variants previously associated with SLC1A4-related disorders are also shown. TM, transmembrane domain. (D) Location of the amino acid Pro91 on an alpha-helix, predicted to destabilize the protein, as seen on HOPE report. (E) Sequence alignment of the residues surrounding Leu91 of human SLC1A4 against other species using the Clustal Omega program.
Later at 6 years of age, neurological examination revealed spastic tetraparesis, with generalized brisk reflexes. Additionally, the patient displayed, jerky movements of the upper limbs.
At the current age of 9 years, he presents severe microcephaly (OFC=47cm, -4.3 SD, lower than 1st percentile) with some dysmorphic features such as hypertelorism, prominent ears, broad flat philtrum, large mouth, thin upper lip and everted lower lip, congenital pectus excavatum and scoliosis. Severe developmental delay is most remarkable. He holds objects for a limited time but does not crawl or sit unsupported. He has consistent eye contact only with caregivers and babbles a limited amount of guttural and monosyllabic sounds. He does not have social laughter and maintains the midline stereotypies with reduced purposeful hand movements. He presents spastic tetraparesis, brisk reflexes in the four limbs, hypersensitivity to touch, irritability and aggressiveness. He maintains disrupted sleep-wake cycles. No hearing or vision impairments were identified. Other clinical features include astigmatism, bruxism, severe osteoporosis and left hip dysplasia which required replacement. Family history was uneventful.
He had a normal EEG and no history of epilepsy.
Comprehensive metabolic screening and electromyogram were normal. CSF studies were not performed.
Brain Magnetic Resonance Imaging (MRI) performed at 10 months of age revealed a thin corpus callosum.
Clinical exome sequencing revealed two heterozygous variants in the SLC1A4 gene, c.527+1G>T and c.1035-1G>C, with possible implications in the phenotype of this patient (Figure 1). Both are novel variants predicted to result in abnormal splicing. Patient parents were not available for segregation analysis, nevertheless, the compatibility with the clinical presentation supports that this patient is a compound heterozygote for the described variants.
To understand the potential effect of the two novel variants on splicing, we performed minigene assays. Minigenes contain the genomic sequence flanking the SLC1A4 variants (Fig. 2), as described in the material and methods section. Analysis of the first variant (c.527+1G>T), showed that expression of the wild-type construct produced a full-length transcript (about 670 bp), while the mutant rendered a main transcript with higher size (about 2890 bp). Sanger sequencing confirmed the latter to retain intron 1 (Figure 2). Therefore, the variant seems to affect splicing by causing the loss of donor splice site of exon 1. Results regarding the second variant (c.1035-1G>C) showed that expression of the wild-type construct produced the canonical transcript (about 620 bp), while the mutant produced two main transcripts (about 620 bp and 750 bp). Sanger sequencing showed that one transcript has a 5 bp deletion in exon 6 and the other a insertion of 19 bp (from intron 5). Therefore, the variant caused the loss of 3’ splice site (acceptor splice site of exon 6), leading to the use of inappropriate 3´splice sites within exon 6 or intron 5 (Figure 2). Given the alteration of splicing, both variants are expected to result in a frameshifts and lead to truncated proteins.
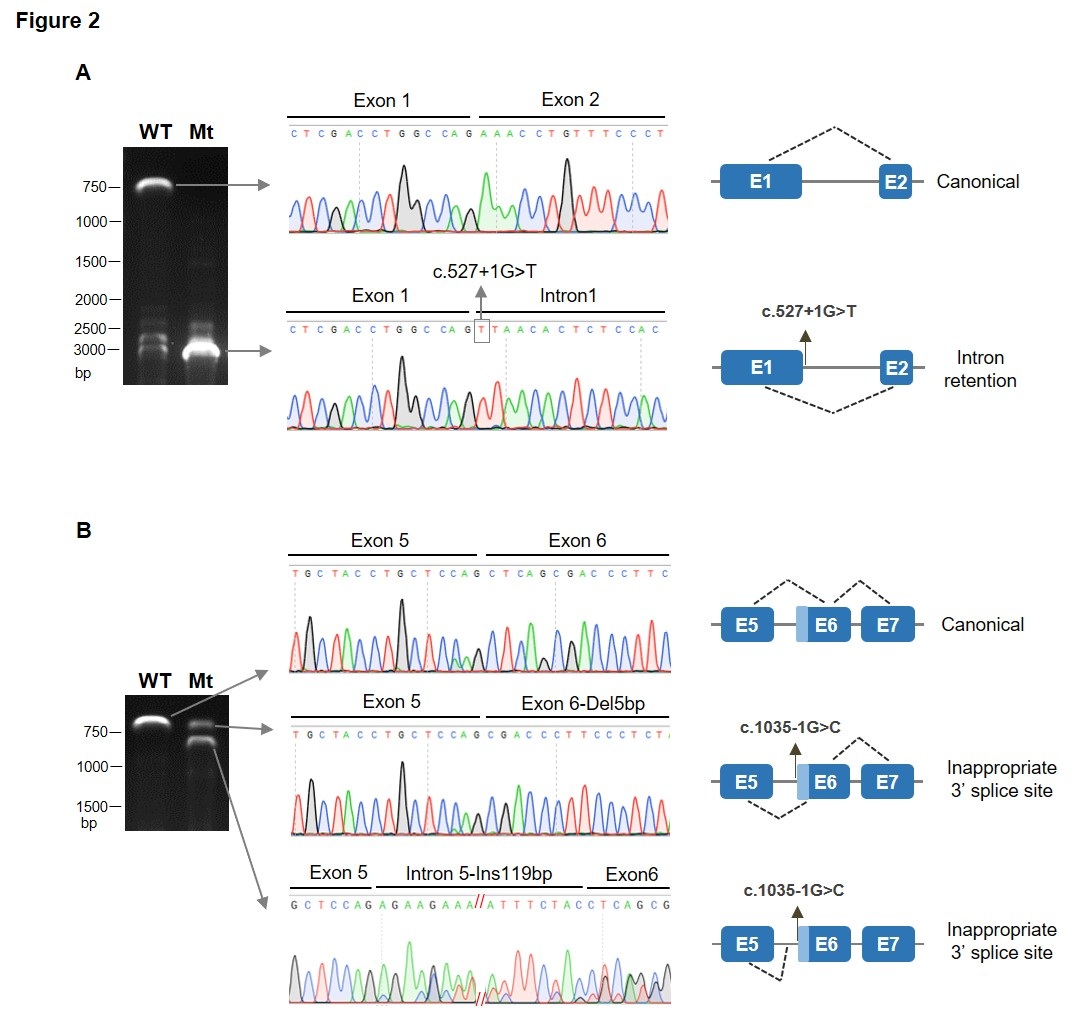
Figure 2: Analysis of SLC1A4 splice site variants by minigene splicing assays. Agarose gel electrophoresis of transcripts generated by the WT and mutant minigene constructs, Sanger sequencing of the corresponding transcripts and schematic representation of resulting splicing events are shown. (A) Analysis of c.527+1G>T variant shows that it causes retention of intron 1. (B) The c.1035-1G>C variant lead to inappropriate 3´splice sites usage within exon 6 or intron 5.
Case 2
The second patient is a 9-year-old girl, only child of consanguineous parents (first cousins) of non-Ashkenazi Jewish ancestry (Figure 1B). She was born at term following an unremarkable pregnancy and delivery.
Patient 2 was normocephalic at birth (OFC=33cm).
At the age of 18 months, she presented bilateral spasticity of the lower limbs, with brisk patellar reflexes and exhaustible ankle clonus on the right. Additionally, she presented a high degree of psychomotor agitation. On repeated examinations, it was noted a severe progressive microcephaly (OFC=45cm/5th percentile at 2 years of age, 47,5cm/2nd percentile at 5 years of age and 49cm/lower the 2nd percentile at 9 years of age). No craniofacial dysmorphic features were noted.
She presented a milder delay when compared to patient 1. At 9 months of age, she could sit unsupported and at 18 months could walk independently with ataxic gate, showing internal rotation of both hips and pes valgus (both more severe on the right side). At 24 months of age, she was verbal and currently, at the age of 9, communicates with two/three-word phrases and attends school with an adapted curriculum, being able to recognize some pictures, colors and doodling. She can feed herself independently. Additionally, she presents hyperkinetic behavior with a lack of concentration on specific tasks, but these characteristics have improved with age and she is becoming more social. She shows brisk reflexes in the lower limbs.
No hearing or vision impairments were identified. Family history was uneventful.
At the age of 5, EEG recording showed epileptogenic activity, namely spike-wave bursts predominantly with a left parieto-temporal focus, evident during sleep, which appears also in the homologous contralateral region in lesser density. Clinical manifestations were not noted but she was medicated for epilepsy. At the age of 8, myoclonus during sleep were reported. In a new EEG, multifocal discharges were described in the front central and frontotemporal regions, either awake and in half of the sleep time.
Comprehensive metabolic screening and electromyogram were normal. CSF studies were not performed.
An MRI performed at the age of 5 was normal without anomalies of the corpus callosum or myelination. aCGH (Microarray-based Comparative Genomic Hybridization) test identified a microdeletion - 20q13.33 that was inherited from her healthy father and classified as probably benign. NGS panel for intellectual disability showed a variant in the gene CDH1 inherited from her healthy mother.
Trio-based WES sequencing was performed and a homozygous variant in SLC1A4 gene c.272T>C (p.(Leu91Pro) was detected (Figure 1B). This substitution replaces a leucine by a proline at position 91, predicted to be probable damaging by different bioinformatic analysis software (PolyPhen-2, SIFT, CADD and MutationTaster). The mutated residue is located within a transmembrane domain (Figure 1C) and it is predicted to disrupt an alpha-helix (Figure 1D), which can have severe effects on the structure of the protein [4]. Moreover, the Leu91 residue of human SLC1A4 is conserved across different species (Fig. 1E). This variant is extremely rare, with only one allele reported, at population databases namely at gnomAD, 1000 Genomes, ESP, or dbSNP. Importantly, this variant has an entry at ClinVar (VCV000801718.1) associated with spastic tetraplegia, thin corpus callosum and progressive microcephaly, and classified as likely pathogenic. Both parents were heterozygotes for this variant.
DISCUSSION
The SLC1A4 related neurological disease (OMIM #616657) represents an extremely rare autosomal recessive neurodevelopmental condition, only first described in 2015, with a high percentage of heterozygous carriers in Ashkenazi-Jewish population (0,7%) for the founder mutation p.E256K. Since then, other pathogenic variants have been described as probable causes for this disorder [5-7]. From a historical point of view, it is important to mention that the Ashkenazi-Jewish population moved between German and Eastern European territories while Iberia received a migration of Sephardi Jews who do not share the same pool of variants [8]. A comparative analysis of the clinical features of all patients with SLC1A4 disease-causing variants reported in the literature, including our two patients, is present in table 1.
Patient 1 had a clinical picture that overlaps with most of the characteristics described by Damseh (Table 1). The hand stereotypies showed by our patient have been reported by Heimer et al., in two cases [6]. Damseh et al., described one patient with a more severe hip dysplasia [5] (Table 1). Epilepsy was not present in patient 1 whereas in patient 2 the EEG showed paroxistic activity. This disparity is consistent with previous reports where only roughly half of the cases presented epilepsy [9,10]. Additionally, patient 1 exhibited, at 3 months of age, progressive microcephaly and midline stereotypies (hand-washing and hand-mouthing) typically observed in females with Rett syndrome and previously reported by Heimer at al., and Abdelrahman et al., [2,6]. His evolution from the initial hypotonia to hypertonia and spasticity is consistent with previous descriptions of children with serine transport defects [11-13].
Patient 2 presents a milder presentation of SPATCCM, with a better functional level when compared not only to patient 1 but also to all patients previously reported with this condition (Table 1). On verbal skills, babbling was present in some patients reported by Damseh et al., and Heimer et al., and one patient, described by Damseh et al. was able to say short sentences as a teenager (Table 1). Regarding locomotion, besides our patient 2, only four patients were reported to walk independently [5-7,14]. The clinical picture of this patient (developmental delay, poor speech and progressive microcephaly) associated with normal MRI, might be related to milder functional effects of the variant found in this patient. However, further MRI studies might show progressive corpus callosum abnormalities.
In both cases WES analysis identified novel SLC1A4 deleterious variants, two heterozygous variants, c.527+1G>T and c.1035-1G>T in patient 1, and a homozygous c.272T>C, p.(Leu91Pro) in patient 2. These variants were found to be extremely rare or absent in several population databases and multiple prediction tools, as well as structural analysis, support their pathogenicity. The p.(Leu91Pro) variant is predicted to disrupt an alpha-helix, which may alter the structure and function of the ASCT1 transporter [4]. Indeed, a previous study showed the p.(Arg457Trp) and p.(Glu256Lys) variants to impair L-serine transport [5]. Additionally, minigene assays for the splice-site variants confirmed their impact in splicing with loss of exon 1 donor splice-site and intron 1 retention, as a result of the c.527+1G>T substitution, and loss of exon 6 acceptor splice-site with inappropriate 3’ splicing sites in exon 6 and intron 5, resulting from the c.1035-1G>T variant. Both variants are expected to result in a frameshift that can either lead to the targeting of the mRNA to nonsense mediated decay or result in a truncated protein.
This notion is further strengthened by the correlation of phenotype severity with the effect of the SLC1A4 disease-causing variant, meaning that a patient who harbors a frameshift variant is more severely affected than the one who is homozygous for a missense variant. The patients here reported lay on distinct extremes of the global development delay spectrum which foresees a spectrum of genotype-phenotype correlations.
SLC1A4 encodes the neutral amino acid transporter ASCT1 (Na+ dependent transporter system ASC), the main L-serine transporter in the brain. L-serine is classified as a non-essential amino acid, although it has a central role in a broad range of cellular processes, including its function as a potent neuronal trophic factor, namely neural survival, growth, differentiation, and dendritic elongation, branching and synaptogenesis by maintaining sphingolipid and phosphatidylserine synthesis. Additionally, it also provides precursors for the synthesis of essential compounds such as amino acids (as L-cysteine), proteins, nucleotides, neuromodulators (as D-serine and glycine), neurotransmitters and L-serine derived lipids [15-17].
L-serine can be derived from different sources: the most important results from diet and the other is de novo biosynthesis from glycolytic intermediate 3-phosphoglycerate. The alternatives, namely direct synthesis from glycine or turnover of proteins and phospholipids, are usually not large enough to compensate for defects in the serine biosynthesis pathway. However, the relative contribution of these sources is still unknown and varies depending on tissue type and developmental stage. Within neural tissues its major source is synthesis de novo, due to limited permeability of the blood-brain barrier for L-serine, behaving almost as an essential amino acid on this tissue [1,12]. In neural tissues, L-serine is synthesized in the astrocytes, from the glycolytic intermediate 3-phosphoglycerate through the sequential catalytic reactions of hydroxyprostaglandin dehydrogenase 15 (15-PGDH), Phosphohydroxythreonine Aminotransferase (PSAT) and Phosphoserine Phosphatase (PSPH). The generated L-serine is then shuttled from its site of synthesis by the dedicated transporter, ASCT1 (with higher affinities for Alanine, Serine and Cysteine) to the extracellular spaces and from there ultimately to its site of action, the neuronal cells.
During the embryonic stage, neural progenitor cells express the biosynthetic enzyme PGDH, which allows the L-serine synthesis and degradation directly on its site of action that is not dependent on transporter ASCT1. This might explain the lack of congenital microcephaly in patients reported to date, with a few exceptions. After birth, neurons lack this enzyme and are dependent on the production of serine on the astrocytes and its effective transport via ASCT1. When the latter fails, L-serine is not able to reach neurons thus compromising its neurotrophic effects, which might explain the progressive microcephaly reported in most patients with SLC1A4 pathogenic variants and also the epileptic manifestations.
CONCLUSION
The report of two patients with new pathogenic variants is important to confirm the role of SLC1A4 in the pathophysiology of unexplained severe neurodevelopmental delay, spasticity and progressive microcephaly, providing further insights for more precise diagnosis of patients with similar clinical features.
It is also important to emphasize that, as aforementioned, the higher carrier rates of a specific disease-causing variant for this disease among the AJ population should not undermine the investigation of SLC1A4 deficiency on any patient with unexplained neurodevelopmental delay and progressive microcephaly, regardless of their ethnic background.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
REFERENCES
- Kaplan E, Zubedat S, Radzishevsky I, Valenta AC, Rechnitz O, et al. (2018) ASCT1 (Slc1a4) transporter is a physiologic regulator of brain D-serine and neurodevelopment. Proc Natl Acad Sci USA 115: 9628-9633.
- Abdelrahman HA, Al-Shamsi A, John A, Ali BR, Al-Gazali L (2019) A Novel SLC1A4 Mutation (p.Y191*) Causes Spastic Tetraplegia, Thin Corpus Callosum, and Progressive Microcephaly (SPATCCM) With Seizure Disorder. Child Neurol Open 6: 2329048X1988064.
- da Glória VG, Martins de Araújo M, Mafalda Santos A, Leal R, de Almeida SF, et al. (2014) T Cell Activation Regulates CD6 Alternative Splicing by Transcription Dynamics and SRSF1. J Immunol 193: 391-399.
- Venselaar H, Te Beek TA, Kuipers RK, Hekkelman ML, Vriend G (2010) Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinformatics 11: 548.
- Damseh N, Simonin A, Jalas C, Picoraro JA, Shaag A, et al. (2015) Mutations in SLC1A4, encoding the brain serine transporter, are associated with developmental delay, microcephaly and hypomyelination. J Med Genet 52: 541-547.
- Heimer G, Marek-Yagel D, Eyal E, Barel O, Oz Levi D, et al (2015) SLC1A4 mutations cause a novel disorder of intellectual disability, progressive microcephaly, spasticity and thin corpus callosum. Clin Genet 88: 327-335.
- Srour M, Hamdan FF, Gan-Or Z, Labuda D, Nassif C, et al. (2015) A homozygous mutation in SLC1A4 in siblings with severe intellectual disability and microcephaly. Clin Genet 88: 1-4.
- Nogueiro I, Teixeira JC, Amorim A, Gusmão L, Alvarez L (2015) Portuguese crypto-Jews: The genetic heritage of a complex history. Front Genet.
- El-Hattab AW (2016) Serine biosynthesis and transport defects. Mol Genet Metab 118: 153-159.
- Pironti E, Salpietro V, Cucinotta F, Granata F, Mormina E, et al. (2018) A novel SLC1A4 homozygous mutation causing congenital microcephaly, epileptic encephalopathy and spastic tetraparesis: a video-EEG and tractography – case study. Journal of Neurogenetics 32: 316-321.
- El-Hattab AW (2016) Serine biosynthesis and transport defects. Mol Genet Metab 118: 153-159.
- Hofmann K, Düker M, Fink T, Lichter P, Stoffel W (1994) Human Neutral Amino Acid Transporter ASCT1: Structure of the Gene (SLC1A4) and Localization to Chromosome 2p13-p15. Genomics 24: 20-26.
- Soma H, Yabe I, Takei A, Fujiki N, Yanagihara T, et al. (2008) Associations between multiple system atrophy and polymorphisms of SLC1A4, SQSTM1, and EIF4EBP1 genes. Mov Disord 23: 1161-1167.
- Conroy J, Allen NM, Gorman K, O'Halloran E, Shahwan A, et al. (2016) Novel European SLC1A4 variant: Infantile spasms and population ancestry analysis. J Hum Genet 61: 761-764.
- Tabatabaie L, Klomp LW, Berger R, de Koning TJ (2010) l-Serine synthesis in the central nervous system: A review on serine deficiency disorders. Mol Genet Metab 99: 256-262.
- Tabatabaie L, de Koning TJ, Geboers AJ, van den Berg IE, Berger R, et al. (2009) Novel mutations in 3-phosphoglycerate dehydrogenase (PHGDH) are distributed throughout the protein and result in altered enzyme kinetics. Hum Mutat 30: 749-756.
- Yamamoto T, Nishizaki I, Nukada T, Kamegaya E, Furuya S, et al. (2004) Functional identification of ASCT1 neutral amino acid transporter as the predominant system for the uptake of L-serine in rat neurons in primary culture. Neurosci Res 49: 101-111.
Citation: Teixeira J, Dória S, Santos M, Alonso I, Leão M (2020) Progressive Microcephaly, Spasticity and Development Delay: Novel SLC1A4 Variants in Two Portuguese Families and Literature Review. J Genet Genomic Sci 4: 017.
Copyright: © 2020 Joana Teixeira, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.