Psoriasis Immunopathogenesis
*Corresponding Author(s):
Zhukov A SDepartment Of Skin And Venereal Diseases, Federal State Budgetary Educational Institution Of Higher Education, Military Medical Academy Named After S.M. Kirov, Ministry Of Defense Of The Russian Federation, Russian Federation
Tel:+7 9818890815,
Email:doctor-vma@mail.ru
Abstract
Psoriasis is a chronic multi-factorial immune-mediated inflammatory disease of skin and joints. The variety of clinical forms of dermatosis is consistent with various pathogenetic features of the disease progress which have been significantly supplemented and reviewed recently. Knowledge of these mechanisms will improve and personalize the prescribed therapy.
This study places the emphasis on modern ideas about the formation of T cell memory, the role of melanocytes and innate lymphoid cells. Development mechanisms of guttate and paradoxical psoriasis with important distinguishing characteristics are described separately.
Today, knowledge of the molecular basis of the disease progression has led to the creation and introduction of a number of highly effective targeted drugs into clinical practice. Further developments related to the inhibition of resident memory cells, innate lymphoid cells, as well as the study of guttate psoriasis perpetuation and the occurrence of paradoxical psoriasis will significantly increase the effectiveness of the therapy.
Keywords
Guttate psoriasis; Innate lymphoid cells; Melanocytes; Paradoxical psoriasis; Pathogenesis; Psoriasis; T-cell memory
Introduction
Psoriasis is a chronic multi-factorial immune-mediated inflammatory disease of the skin. According to the World Health Organization, about 125 million out of more than 7.7 billion of the world population suffer from psoriasis. The prevalence of psoriasis among the population varies widely: from 0.09% in Tanzania to 5.1% in the United States which is 1-3% of the world population on the average. Men and women suffer from psoriasis equally [1-3].
This report focuses on the issues of predisposition to disease development, the features of clinical findings and pathomorphology, the participation of T cell subpopulations in the inflammatory response, the formation of T cell memory, the role of dendritic cells, melanocytes and innate lymphoid cells. Distinctive characteristics of the pathogenesis of guttate and paradoxical psoriasis that affect the case management strategy are described separately.
Underlying Risk for Disease Development
Major genetic studies have shown that the underlying risk for psoriasis is inherited poligenously and is associated with genes that manage adaptive and congenital immune responses and epidermal barrier functions [4,5]. Moreover, the association between the Cw6 allele of the major Histocompatibility Complex (HLA-C) gene and underlying risk for plaque psoriasis, as well as the early disease onset remain the most reproducible in different ethnic groups [6-8]. The probability of the disease development in one of the monoovular twins, the second of which has fallen with psoriasis, is 60-75% [9].
The factors that provoke psoriasis development and worsen its clinical course include lymphoid periopharyngeal ring streptococcal infection, psychological stress, smoking, excessive alcohol consumption, use of certain medications (β-adrenergic blocking agents, α-interferon, aminoquinolines, etc.) [10-12].
Clinical, histopathological, and immunohistochemical states of psoriasis are heterogeneous, variable, and need to be studied and compared in detail to understand the disease development mechanisms.
Clinical Features of Psoriasis
The difficulties in studying psoriasis pathogenesis are considerably related to different clinical findings and disease progression. For example, plaque psoriasis is characterized by plane pink papules that have a propensity for the peripheral growth forming plaques, pustular psoriasis is characterized by amicrobic pustules, psoriatic erythroderma is characterized by “edematous spot” and exfoliative peeling, and psoriatic arthritis is characterized by inflammation of peripheral joints, fingers, entheses, vertebral column, and iliosacral pelvic joints. Psoriasis may be either a mild disease characterized by individual insignificant plaques during a long period of time or severe disease that affects a large area of the skin and characterized by arthritis mutilans development and the significant decrease in the patients' life quality.
Such heterogeneity of clinical implications can be explained by psoriasis polygenetic nature. Various combinations of different predisposing genes of a patient create individual psoriasis models in each patient; those models differ in phenotypic implications, the disease severity, and the therapy effectiveness. At the same time, identical twins with psoriasis often have similarities in clinical findings of the disease, the age of the disease onset, the nature of the disease progression, the presence or absence of joint damage [13-16].
Autoimmune Inflammation
The autoimmune process in psoriasis is deemed one of the major mechanisms of disease development. It is expected that keratinocytes are trigger cells. In the event of a damage (trauma, infections, drugs, UV), they "supply" autoimmune antigens and activate antigen-presenting cells due to the secretion of a large set of congenital immunity factors (cytokines, chemokines, antimicrobial peptides) [17,18].
Various compounds are considered probable autoimmune antigens: cathelicidin (LL-37) and β-defensins from the group of antimicrobial peptides; neolipid antigens produced by mast cell phospholipase; K16 and K17 keratins that are homologous to M protein of streptococcus, and the cellular antigen (ADAMTSL5 protein located in melanocytes) [19-23].
The key moment in the autoimmune inflammation initiation is the immune tolerance failure which is currently associated with the activity of cytosolic and extracellular DNA. Normally, a DNA molecule in the cell is only in the nucleus, small fragments are present in the mitochondria. DNA may be found in the cytoplasm at pathologically increased permeability of the nuclear membrane, mechanical or immune damage, and uptake of neighboring destroyed cells DNA by keratinocytes. In this case, when DNA enters the cytoplasm of keratinocytes, it stimulates the interleukine production (IL)-1β through interaction with various intracellular DNA sensors (protein AIM2 and others). In other cases, when forming stable complexes with antimicrobial peptides (cathelicidin LL37, beta-defensin (hBD) 2, hBD3 and lysozyme), extracellular nucleic acid fragments (DNA and RNA) acquire the ability to transport themselves through cell membranes to endosomal compartments with TLR7 and TLR9; it leads to the activation of Plasmacytoid Dendritic Cells (pDCs) followed by secretion of I and II type interferons (INF) [24-28]. IL-1β and α-INF are important molecules that enhance the expression of HLA antigens, activation, and maturation of dendritic cells; therefore, they contribute to the immune tolerance loss.
Repeated location of the psoriatic eruption on the hairy part of the head, elbow, and knee joints may be connected with traumatization of these skin areas and damage of keratinocytes that leads to DNA yield into the cytosol. Isomorphic reaction development is explained in a similar way.
Role Of T Lymphocytes
The introduction of an immunohistochemical study of the affected skin of psoriasis patients allowed scientists to determine the phenotype of the main immune cells involved in the inflammatory process. One of the dominant cell populations are T lymphocytes (CD3+), the number of which in the eruption areas increases by 6-10 times compared with healthy skin. About 2/3 of T lymphocytes are represented by CD4+-cells which are found exclusively in the dermis (they are located in groups (nests)), and cytotoxic T lymphocytes (CD8+) which are scattered: about 1/4 of them are found in the epidermis, and 3/4 are found in dermal papillae. CD4+-lymphocytes consist of subpopulations of 1 and 17 type T helpers (Th1 u Th17), and T regulatory cells (T reg). T regulatory lymphocytes make up about 20-25% of all T cells and are found mainly in dermal papillae as part of infiltrates. More than 90% of all T lymphocytes have a CD45RO molecule on their surface, which indicates the maturity of these cells and their transportation in the lymphoid organs of an antigen-specific differentiation.
Th1 lymphocytes produce a variety of proinflammatory cytokines in the skin of psoriasis patients; γ-interferon (γ-IFN) is the key one. The role of Th1 cells at the earliest inflammation stages is to strengthen and expand the boundaries of this process, involving other participating cells in it. By synthesizing γ-IFN which controls the transcription of a group of interferon-stimulated genes, Th1 induces enhanced synthesis of neutrophil-activating factors, adhesion molecules, pro-inflammatory cytokines, and other biologically active substances by surrounding cells (keratinocytes, fibroblasts, endotheliocytes, etc.). Due to neutrophil-activating factors (CCR5, CXCL9, CXCL10, IL-8), a significant number of immune cells are concentrated in the area of the forming psoriatic papule which is represented mainly by various subpopulations of T lymphocytes, DCs, monocytes, and neutrophilic leukocytes. The secreted Th1 cytokines TNF-α and IL-6 which have diverse effects are the most important mediators of the acute inflammation phase [29,30].
CD8+-lymphocytes of psoriasis patients interact with antigen-presenting DCs in lymphoid organs and skin and stimulate their production of IL-12 (IL-12p70 subunits). The secretion of this cytokine causes the differentiation of naive T cells in Th1, significantly increases their number, and suppresses Th2 formation. Another important function of CD8+ cells in forming psoriatic plaques is their participation in the rapid differentiation of monocytes which migrate into the affected skin from peripheral blood, into CD11c+ DCs [31,32].
Role of T Helpers of 17 Types
Th17-lymphocytes are practically not found in healthy skin. The participation of these cells in the inflammatory process is associated with the autoimmune response development [33]. Th17 are major figures in psoriasis pathogenesis, their participation in the inflammatory process gives it a specific character and leads to the development of clinical implications specifically attributed to psoriasis. Global genetic studies have revealed an association between psoriasis development and the polymorphism of IL23R and TRAF3IP2 genes which regulate the number and activity of Th17 subpopulation [34,35]. The experience in the application of antibodies that block the common IL12/IL23 subunit, IL17 cytokine, or its IL17R receptor has demonstrated its high efficiency in psoriasis therapy [36-38]. Th17 express RORC transcription factor, receptor to IL23, CCR6 chemokine receptor, and receptor to lectin CD161. The differentiation in Th17 comes from precursors - CD4+CD161+-cells in the presence of IL-1β and IL-23. Mature Th17 produce anti-inflammatory cytokine; IL-17 and IL-22 interleukins are important. IL-17 that consists of IL-17A and IL-17F monomers can connect with IL-17 receptor which expresses itself on keratinocytes, endotheliocytes, T lymphocytes, monocytes, fibroblasts. Such interaction results in the production of IL-6 and IL-8 cytokines. The secretion of IL-8 which is a chemo attractive agent for neutrophilic leukocytes causes the accumulation of such cells in the area of psoriatic eruption and forms Munro's micro abscesses [39-44]. IL-17 and IL-22 cause hyper proliferation and keratinocyte differentiation impairment which leads to the development of epidermal hyperplasia, agranulosis, and hyperparakeratosis [45,46]. IL-17 also causes the expression of a variety of chemokines that involve T lymphocytes, monocytes, and DCs into the inflammation site. CXCL13 and CCL19 chemokines appear on fibroblasts under the influence of IL-17. These chemokines lead to migration of lymphocytes to the dermis and can induce the formation of ectopic lymphoid tissue, where new autologous-reactive T lymphocytes are formed [47,48].
Formation of T Cell Memory at Psoriasis
T lymphocytes that have passed special differentiation pass from lymphoid organs into the skin and joints (at patients with psoriatic arthritis) due to the trafficking of these cells provided by chemokines. As soon as the inflammation is ended and psoriatic eruption disappears, a part of these lymphocytes stays in the skin and forms immune memory. Such cells which store information about a certain antigen (resident memory T cells, TRM) are an integral part of the adaptive immunity.
The biological sense of forming the anamnesis lies in the development of a more rapid immune response to repeated contact of a memory cell with a known antigen. Activation of memory cells requires lower doses of antigen than for differentiation of naive T lymphocytes [49,50]. Memory T cells are present in all peripheral tissues of a healthy person, including skin where their share is 95% of all lymphocytes. There are 2 times more memory T cells in the skin than in peripheral blood. These cells move at the border with the external environment and constantly screen pathogens. In the case of anti-infectious and anti-tumor defense, TRM undoubtedly play a positive role. However, the formation of TRM in response to allergens or autoimmune antigens becomes a serious problem for the body [51-53].
Psoriasis is a chronic immune-related dermatosis, and the formation of a T cell immune response that develops autoimmune inflammation plays the main role in its pathogenesis. The preservation of memory T cells after psoriasis onset is the formation of the disease's immunological memory; they explain its incurability and relapsing course. Memory T cells are presented by long-lived populations of central (TCM) and resident T lymphocytes. TRM are mostly located in the skin, they do not move to the systemic blood and have the phenotype CD45RO+CCR7−CD69+CD103+ [54-58]. The further study of TRM’s subpopulation pattern in psoriatic eruptions has detected the predominance of Th22 cells (CD4+-cells that produce IL-22) and Tc17 (CD8+-cells that produce IL-22) which form the memory about the disease and can provoke the disease recrudescence in case of their stimulation [59-61].
The appearance of eruptions at psoriasis recrudescence can occur only with the participation of dermal TRM s without involving lymphocytes from lymphoid organs. This fact may be proved by studies on the application of E-selectin inhibitors at research animals. E-selectin antibodies hinder the migration of T cells from blood to the skin. Clinical studies at immune compromised mice have shown that after the introduction of E-selectin inhibitors and transplantation of clinically unchanged skin of psoriasis patients, psoriatic eruptions have developed spontaneously or in case of stimulation with TNFα [62-63].
With the development of psoriasis recrudescence, T cell proliferation is observed in the dermis (in the psoriatic papule). Immunohistological analysis of the skin of psoriasis patients during the disease progression using the double staining method for CD3ε and Ki67 has allowed to find out that about 30% of all T lymphocytes in dermal infiltrates have been positive for both markers at the same time, i.e. they have been in the stage of mitosis. Therefore, the formation of inflammatory infiltrates in the foundations of psoriatic papules may be caused by an intradermal proliferation of T cells [64].
Studies of the 1st phase of the drug which inhibit resident memory T cells in psoriasis patients have already been completed and the first positive results have been obtained [65].
Role of Innate Lymphoid Cells
The last scientific findings also point to the important role of Innate Lymphoid Cells (ILCs) in psoriasis pathogenesis [66,67]. ILCs are characterized by the expression of a molecule of leukocyte common antigen (CD45), α-chains of IL-7 receptor (CD127) and the absence of specific markers of DCs, T, and B-lymphocytes including the engineered receptor for recognizing the antigen [68,69].
ILCs family may be divided into three groups according to their need in activation with cytokines, expression of transcription factors, and production of effector cytokines. ILCs of the 1st group include NK-cells and ILC1 which are activated by IL-12, depend on T bet, and produce IFN-γ. The 2nd group includes ILC2 which are activated by L-25, IL-33, and TSLP, express GATA-3 and produce IL-4, IL-5, and IL-13. ILCs of the 3rd group include LT is (lymphoid tissue inducer cells) and ILC3. They are activated by IL-1 and IL-23, depend on RORγt and produce IL-17A and/or IL-22. ILC3 is further subdivided based on the expression of Natural Cytotoxicity Receptors (NCR). Interestingly, all activating cytokines may be produced by keratinocytes, Langerhans cells, and DCs of the skin [68,70].
All three subsets of ILCs are present in the healthy skin and may functionally (according to the production of cytokines) match with adaptive response phenotypes mediated by T lymphocyte helpers (ILC1 (Th1), ILC2 (Th2) and ILC3 (Th17)). It is acknowledged that ILCs are important regulators of tissue homeostasis and inflammation [69,71,72].
The first report on the possible role of ILC3 in psoriasis development has been made by Pantelyushin S. et al. in 2012. They have shown that RORγt+ILC3 and γδT cells have been the main cells producing IL-17 and IL-22 in the experimental psoriasis model in mice [73]. Subsequently, several studies have confirmed the primary outcome. For example, Brüggen MC, et al. have performed a comparative analysis of ILCs subsets in the skin of patients with psoriasis, atopic dermatitis, and skin of healthy people and have revealed the predominance of ILC3 and ILC1 in psoriatic foci, while the main population is ILC2 at atopic dermatitis. Moreover, topographic mapping of ILCs using in situ IF staining has shown the predominant location of ILCs near the epidermis and in close proximity to T lymphocytes, which suggests a direct interaction between these cell types [71]. The other study by Dyring-Andersen B, et al. has shown the increase in the number of RORγt+CD56+ILC3 producing IL-22 in both affected and unaffected skin of psoriasis patients in comparison with the skin of healthy persons and patients with nickel contact allergy [74].
Two more studies have revealed the significant increase in NCR+ILC3 at psoriasis patients not only in the skin but also in the peripheral blood. After in vitro stimulation of IL-1 and IL-23, NCR+ILC3 derived from psoriatic foci have mainly produced IL-22 and IL-17 (fewer). Significant differences have been obtained in comparing the amount of NCR+ILC3 in the unaffected skin of psoriasis patients and healthy persons. This fact points to the amended allocation of ILCs in the skin of psoriasis patients which may be a predisposing or initiating factor for dermatosis development [69,75].
The study using the humanized mouse model of psoriasis has proved that ILC3 cells not only increase quantitatively in the foci of the affected skin but can also lead to the development of a psoriatic phenotype even in the absence of Th17 cells [76].
Thus, ILC3 can make a significant contribution to psoriasis pathogenesis by producing key anti-inflammatory IL-17 and IL-22 cytokines. Therefore, ILCs modulation is a new therapeutic approach to psoriasis patient management in the future.
Role Of Dendritic Cells
Dendritic Cells (DCs) are presented in the skin of psoriasis patients by single plasmacytoid DCs (pDCs) which express CD123+ and a large population of myeloid DCs. The latter include several varieties of DCs. These are Langerhans cells (young DCs, Langerin+, CD1a+) which are located in the lower epidermis layers at healthy persons and psoriasis patients during the remission; they form a chain by connecting with their processes. A significant number of them can be found in the dermal papillae during the progressing period of the disease. CD11c+-cells (TNF-α/iNOS-producing DCs) are localized mostly in dermal in filtrates, their number is equal to all T lymphocytes. About 10% of myeloid DCs are mature CD83+-cells located in the dermis (near epidermal and dermal linkage) and the epidermis.
Langerhans cells play a key role in the formation of the immune response; they uptake foreign antigens/autoimmune antigens, process them along with II class MHC molecules, and if an activation signal has been obtained from keratinocyte, they move to the dermis where they involve pDCs into the inflammation. pDCs are found in the skin of psoriasis patients mainly in the initial stages of the inflammatory process. These cells secrete significant numbers of α- u β-IFN which make the migration of T lymphocytes to the forming psoriatic papule [77].
The activation of Langerhans cells after moving to the dermis is accompanied by their differentiation into dermal myeloid DCs, a change in the immunophenotype (a loss of the specific marker CD207+/Langerin+ is observed), and a loss of phagocytizing properties. Due to the expression of the CCR7 chemokine receptor, such DCs migrate to the regional lymphoid organ (lymphoid ring, lymph nodes) where they participate in the antigen-presenting differentiation of naive T lymphocytes as antigen-presenting cell and ensure the development of the adaptive immune response [78].
CD11c+-DCs represent one of the most numerous populations found in the skin of psoriasis patients. Most monocytes migrate to the dermis, are accumulated in the foundation of the psoriatic papule, and are differentiated in CD11c+-DCs. The increase in the number of CD11c+-cells at psoriasis patients is observed only in the skin, not peripheral blood; this shows their intradermal differentiation. These cells are the main source of the inflammatory key cytokine synthesis (TNF-α) and inducible Nitrogen Oxide Synthase (iNOS). That is why they are called TNF-α/iNOS-producing DCs (Tip-DCs). INOS ferment causes pronounced vasodilation of the dermal vessels in psoriatic eruptions by forming nitrogen oxide [79-81]. CD11 molecule is the integrin that performs the cellular interaction at B- and T cell proliferation. When treating psoriasis patients with efalizumab (CD11a-antibodies, Raptiva), a significant decrease in CD11c+-DCS has been noted in the skin. Moreover, the decrease in CD11c+-cells has been preceded by a decrease in T lymphocytes' amount in the infiltrate and the keratinocyte proliferative rate normalization. The population dynamics of CD11c+-cells has had the best correlation with the clinical response to anti-cytokine therapy [82].
Participation Of Melanocytes In Psoriasis Development
When studying the skin of psoriasis patients, Arakawa A, et al. (2015) have found that a major part (37%) of cytotoxic lymphocytes (CD8+) directly contact with epidermal melanocytes [19]. It is also established that psoriatic plaques have an increased number of melanocytes [83]. Assuming that melanocytes are the target of autoimmune inflammation at psoriasis, Arakawa A, et al. have studied the antigens presented in these cells and have found a possible autoimmune antigen. They have found ADAMTSL5 melanocytic protein which has formed an auto-aggressive clone of CD8+ cells (together with HLA-C*0602 antigen) and has stimulated them to produce IL17 (the so-called Tc17-cells) initiating psoriatic inflammation in the skin. The researchers have noted that the HLA-C*0602 allele is involved in the development of the autoimmune response against melanocytes, so the carriers of this antigen have a huge risk of psoriasis development [19].
The results of our studies have also confirmed that the absolute number of melanocytes in the affected skin of psoriasis patients has been significantly higher than in the unaffected skin and skin of healthy persons, while the ratio between melanocytes and basal keratinocytes is the same (Figure 1). It is important to note that no melanocytes in the proliferating stage have been found in all groups except for 1-2 cases (Figure 2). Thus, we have reached the conclusion that the increased number of melanocytes is not connected with mitosis in the epidermis but probably occurs in the hair follicle bulge where the germ-line cells are located. Figure 3 shows multiple contacts of MelanA+ and CD8+ cells in the affected skin of psoriasis patients which may be indicative of the immune interaction between these cells.
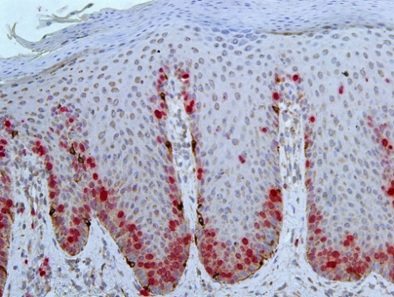 Figure 1: Immunohistochemical study (dual detection system): neutral redstaining - marker Ki-67, Bismarck brown staining - marker MelanA); magn: ×200, 696×507 μm.
Figure 1: Immunohistochemical study (dual detection system): neutral redstaining - marker Ki-67, Bismarck brown staining - marker MelanA); magn: ×200, 696×507 μm.
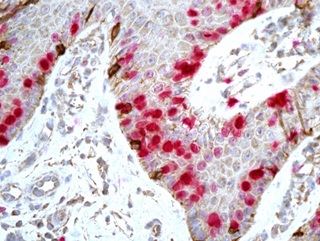 Figure 2: Immunohistochemical study (dual detection system). Sporadic proliferating melanocyte (MelanA+-Ki67+-cell) in the epidermis of psoriasis patients (marked by a narrow); magn. ×400, 348×253 μm.
Figure 2: Immunohistochemical study (dual detection system). Sporadic proliferating melanocyte (MelanA+-Ki67+-cell) in the epidermis of psoriasis patients (marked by a narrow); magn. ×400, 348×253 μm.
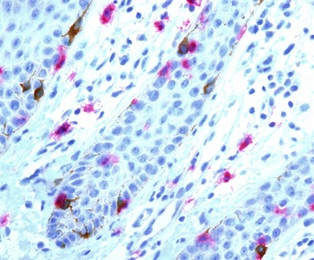 Figure 3: Immunohistochemical study (dual detection system). The contacts ofMelanA+ cells (Bismarck brown staining) and CD8+ cells (neutral red staining) in the epidermis (marked by arrows); magn. ×400, 348×253 μm.
Figure 3: Immunohistochemical study (dual detection system). The contacts ofMelanA+ cells (Bismarck brown staining) and CD8+ cells (neutral red staining) in the epidermis (marked by arrows); magn. ×400, 348×253 μm.
However, it is not understood how psoriatic eruptions appear on the depigmented skin areas of patients with acquired leukoderma which do not have melanocytes. Also, pre-melanocytes (which are the predecessors of melanocytes) located in the hair follicle (bulge area) which may take part in the formation of eruptions in depigmented areas have not been studied to date.
We should describe more specifically two forms of psoriasis: guttate psoriasis and paradoxical psoriasis. It is related to the anormogenesis and importance of their study for understanding the disease as a whole. Guttate psoriasis is often the first stage of the disease, and the detection of a mechanism of its transformation into the chronic plaque form has an important prognostic significance. Paradoxical psoriasis occurs at treatment with genetically engineered biologic drugs, and the explanation of this phenomenon can help to revise views towards key effect points at psoriasis treatment.
Guttate Psoriasis Development Mechanism
This form is generally associated with the presence of focal infection in the form of Chronic Tonsillitis (CT). According to our information, 85.0% of GP patients have associated dermatosis development or recrudescence with CT recrudescence’s, while a similar relationship was observed only in 22.4% of cases with plaque psoriasis. Today, Streptococcus pyogenes is the key factor of guttate psoriasis pathogenesis; the infection's main reservoir is located in palatine tonsils.
The interaction of several factors that lead to the development of a specific clinical pattern of GP (tiny disseminated mildly-infiltrated papules which do not have a propensity to peripheral growth) is supposed. Firstly, the expression of a Cutaneous Lymphocyte Antigen (CLA) molecule is induced under the influence of streptococcus superantigens on the surface of lymphocyte’s CD4+ and CD8+ tonsils formed in T areas, which means that they are able to penetrate the skin [84]. Secondly, the antigen mimicry mechanism is triggered. In this case, T cells can interact not only with peptides of M6 streptococcal protein but also with homologous keratine sequences in the skin [85,86]. Thirdly, blood monocytes uptake the destroyed sacculus components coming out of the inflammation focus (M-proteins, peptidoglycan), migrate to the skin, differentiate to immature DCs, and activate lymphocytes which infiltrate dermis (CLA+CD4+) and epidermis (CLA+CD8+) [87-89]. CD8+ T-cells trigger the keratinocyte proliferation, and CD4+ T-cells support immune inflammation [1]. It is important to recognize that over time, as a result of the cross-presentation of the released autoimmune antigens, it is possible to switch the immune response from the tonsil axis→skin, to the skin axis↔regional lymph nodes which will result in a decrease in the eruption relapse dependence from CT recrudescence and the transition of the process into a plaque form of dermatosis.
This hypothesis is based on the following scientific findings. Psoriasis patients have an increased number of CD4+ and CD8+ T-lymphocytes in palatine tonsils and peripheral blood which express the CLA targeted molecule on their surfaces. Moreover, a high level of receptor expression to Interleukine-23 (IL) which plays a key role in their differentiation in Th17 is found at psoriasis patients on CLA-positive T lymphocytes of tonsils [84]. These data clearly demonstrate the fact that pharyngeal lymphoid tissue ring (secondary lymphoid organ) is a source of effector T lymphocytes that migrate to the skin (resident dermal T lymphocytes) at psoriasis. The inflammatory process in the pharynx lymphoid tissue often caused by Streptococcus pyogenes only stimulates the intensive proliferation and differentiation of T cells, their release into the systemic circulation. The hematogenous spread of these lymphocytes can be inferred by the disseminated nature of eruptions with this psoriasis form.
Paradoxical Psoriasis Development Mechanism
Psoriasis development mechanism during treatment with Genetically Engineered Biologic Drugs (GEBD) is not fully understood. Initially, some authors have considered psoriasiform eruptions as a delayed-type hypersensitivity reaction on the skin, but histological studies have demonstrated the identity of pathomorphological changes in patients with paradoxical and ordinary psoriasis [90].
The most possible is the hypothesis of the development of a disbalance between TNF-α and INF-α cytokines in patients who take genetically engineered biologic drugs. TNF-α inhibits activity and maturation of pDCs which are the main sources of INF-α. These cells appear in the dermis at the early stages of psoriatic papule formation, and it appears that they take part in the immune inflammation initiation. The secreted pDCs of INF-α lead to the increase of CXCR3 expression which causes the migration of Th2 auto responsive cells. The transfer of the inflammatory process in the skin is accompanied by an increase of TNF-α concentration in psoriatic papules and INF-α synthesis depression. The application of TNF-α inhibitors may lead to activation of pDCs and excessive production of INF-α which triggers the psoriatic inflammation in the skin [91,92].
The participation of TNF-α in the regulation of the interaction between T effector and T regulatory cells in the inflammatory process has been reviewed in experimental studies by Chen X, et al. (2007). They have demonstrated on cell cultures consisting of CD4+CD25– T lymphocytes and T regulatory cells that the short term (<48 hours) exposure of TNFα leads to depression of T regulatory cells' suppressor effect upon the proliferation of T effectors. A longer-term presence of TNFα has been accompanied by the suppressor activity restoration in T regulatory cells, cytokine secretion suppression, and CD4+CD25-T lymphocyte proliferation. The authors have assumed that the physiological implication of this phenomenon is in the suppression of immunosuppressive action of T regulatory cells in order to initiate the inflammatory process by T effectors with the subsequent immune response inhibition [93]. The TNFα has a direct effect on T cells through the TNFRII receptor expressed on their surfaces. The application of anti-TNFRII monoclonal antibodies leads to a decrease in the population of T regulatory cells in psoriatic eruptions [94]. From this point of view, we can try to explain the paradox when psoriasis has manifested in patients or psoriatic arthritis patients who have had psoriatic eruptions on the skin during the treatment of rheumatoid arthritis with monoclonal antibodies to TNFα [95]. In these cases, the neutralization of the TNFα stimulatory effect must have been accompanied by the suppression of T regulatory cells that have led to the psoriatic inflammation initiation in the skin.
Conclusion
Since the last century, our visions of psoriasis pathogenesis have changed a lot. The concept of the immune system malfunctions has replaced the prevailing theory of the key role of a primary abnormality of keratinocyte differentiation. It is assumed that the balance change towards the increased synthesis of pro-inflammatory cytokines by immune-competent cells leads to the development of psoriatic manifestations in the skin.
This review presents the main components of psoriasis pathogenesis based on the disease development immune theory. The immune system cells which participate in the formation of a psoriatic phenotype are described. Recent studies on the role of melanocytes, congenital lymphoid cells, and T cell memory in the disease development are highlighted. The emphasis is on the fact that the autoimmune inflammation is the guide link explaining the pathogenesis of this dermatosis. At the same time, no auto reactive antibodies have been detected up to the present, and the autoimmune antigens proposed to researchers are either underexplored or are localized in other organs and tissues where the inflammation does not develop.
The necessity for a detailed study of a focal infection during guttate and plaque psoriasis is of special interest. Despite the fact that there is no large-scale study that proves the significance of the chronic infection foci in plaque psoriasis formation risk up to the present, most researchers have experimentally established a positive effect of indolent inflammatory disease sanation in regression and a more favorable dermatosis course. However, it is not understood why the disease course becomes chronic in only a third of patients with guttate psoriasis and what factors contribute to it.
Today, the study of the molecular basis of the disease progression has led to the creation and introduction of a number of highly effective targeted drugs into clinical practice. Further developments related to the inhibition of resident memory cells, innate lymphoid cells, as well as the study of guttate psoriasis perpetuation will significantly increase the effectiveness of the therapy.
References
- Parisi R, Symmons DP, Griffiths CE, Ashcroft DM (2013) Identification and Management of Psoriasis and Associated ComorbidiTy (IMPACT) project team. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 133: 377-385.
- Michalek IM, Loring B, John SM (2017) A systematic review of worldwide epidemiology of psoriasis. J Eur Acad Dermatol Venereol 31: 205-212.
- Boehncke WH, Sch?¶n MP (2015) Psoriasis. Lancet 386: 983-994.
- Tsoi LC, Spain SL, Knight J, Ellinghaus E, Stuart PE, et al. (2012) Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat Genet 44: 1341-1348.
- Tang H, Jin X, Li Y, Jiang H, Tang X, et al. (2014) A large-scale screen for coding variants predisposing to psoriasis. Nat Genet 46: 45-50.
- Chandra A, Ray A, Senapati S, Chatterjee R (2015) Genetic and epigenetic basis of psoriasis pathogenesis. Mol Immunol 64: 313-323.
- Gudjonsson JE, Karason A, Antonsdottir A, Runarsdottir EH, Hauksson VB, et al. (2003) Psoriasis patients who are homozygous for the HLA-Cw*0602 allele have a 2.5-fold increased risk of developing psoriasis compared with Cw6 heterozygotes. Br J Dermatol 148: 233-235.
- Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NVC, et al. (2006) Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet 78: 827-851.
- Lonnberg AS, Skov L, Skytthe A, Kyvik KO, Pedersen OB, et al. (2013) Heritability of psoriasis in a large twin sample. Br J Dermatol 169: 412-416.
- Weigle N, McBane S (2013) Psoriasis. Am Fam Physician 87: 626-633.
- Kimball AB, Leonardi C, Stahle M, Gulliver W, Chevrier M, et al. (2014) Demography, baseline disease characteristics and treatment history of patients with psoriasis enrolled in a multicentre, prospective, disease-based registry (PSOLAR). Br J Dermatol 171: 137-147.
- Mendieta KL, Irfan M, Fernandez Faith E (2018) Interferon-alpha induced psoriasis in a teenager. Pediatr Dermatol 35: 136-137.
- Farber EM, Nall ML, Watson W (1974) Natural history of psoriasis in 61 twin pairs. Arch Dermatol 109: 207-211.
- Brandrup F, Hauge M, Henningsen K, Eriksen B (1978) Psoriasis in an unselected series of twins. Arch Dermatol 114: 874-878.
- Mori N, Yoshikawa K, Ohno M (1980) Psoriasis occuring in young monozygotic twins. J Dermatol. 7: 71-73.
- L?ennberg AS, Skov L, Skytthe A, Kyvik KO, Pedersen OB, et al. (2013) Heritability of psoriasis in a large twin sample. Br J Dermatol 169: 412-416.
- Nestle FO (2009) Skin immune sentinels in health and disease. Nat Rev Immunol 9: 679-691.
- Albanesi C, Madonna S, Gisondi P, Girolomoni G (2018) The Interplay Between Keratinocytes and Immune Cells in the Pathogenesis of Psoriasis. Front Immunol 9:1549.
- Arakawa A, Siewert K, Stöhr J, Besgen P, Kim SM, et al. (2015) Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med 212: 2203-2212.
- Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, et al. (2014) The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun 3: 5621.
- Cheung KL, Jarrett R, Subramaniam S, Salimi M, Gutowska-Owsiak D, et al. (2016) Psoriatic T cells recognize neolipid antigens generated by mast cell phospholipase delivered by exosomes and presented by CD1a. J Exp Med 213: 2399-2412.
- Fuentes-Duculan J, Bonifacio KM, Hawkes JE, Kunjravia N, Cueto I, et al. (2017) Autoantigens ADAMTSL5 and LL37 are significantly upregulated in active psoriasis and localized with keratinocytes, dendritic cells and other leukocytes. Exp Dermatol 26: 1075-1082.
- Johnston A, Gudjonsson JE, Sigmundsdottir H, Love TJ, Valdimarsson H (2004) Peripheral blood T cell responses to keratin peptides that share sequences with streptococcal M proteins are largely restricted to skin-homing CD8(+) T cells. Clin Exp Immunol 138: 83-93.
- Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, et al. (2007) Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature 449: 564-569.
- Kahlenberg JM, Kaplan MJ (2013) Little peptide, big effects: the role of LL-37 in inflammation and autoimmune disease. J Immunol 191: 4895-4901.
- Lande R, Chamilos G, Ganguly D, Demaria O, Frasca L, et al. (2015) Cationic antimicrobial peptides in psoriatic skin cooperate to break innate tolerance to self-DNA. Eur J Immunol 45: 203-213.
- Dombrowski Y, Peric M, Koglin S, Kammerbauer C, Göss C, et al. (2011) Cytosolic DNA Triggers Inflammasome Activation in Keratinocytes in Psoriatic Lesions. Sci Transl Med 3: 8238.
- Paludan SR, Bowie AG (2013) Immune sensing of DNA. Immunity 38: 870-880.
- Chiliveru S, Rahbek SH, Jensen SK, Jørgensen SE, Nissen SK, et al. (2014) Inflammatory cytokines break down intrinsic immunological tolerance of human primary keratinocytes to cytosolic DNA. J Immunol. 192: 2395-2404.
- Campanati A, Orciani M, Consales V, Lazzarini R, Ganzetti G, et al. (2014) Characterization and profiling of immunomodulatory genes in resident mesenchymal stem cells reflect the Th1-Th17/Th2 imbalance of psoriasis. Arch Dermatol Res 306: 915-920.
- Wong K, Lew F, MacAry P, Kemeny D (2008) CD40L-expressing CD8 T cells prime CD8alpha(+) DC for IL-12p70 production. Eur J Immunol 38: 2251-2262.
- Chong SZ, Wong KL, Lin G, Yang CM, Wong SC, et al. (2011) Human CD8+ T cells drive Th1 responses through the differentiation of TNF/iNOS-producing dendritic cells. Eur J Immunol 41: 1639-1651.
- Fouser LA, Wright JF, Dunussi-Joannopoulos K, Collins M (2008) Th17 cytokines and their emerging roles in inflammation and autoimmunity. Immunol Rev 226: 87-102.
- Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, et al. (2007) A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet 80: 273-290.
- Ellinghaus E, Ellinghaus D, Stuart P, Nair RP, Debrus S, et al. (2010) Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet 42: 991-995.
- Tan JY, Li S, Yang K, Ma B, Chen W, et al. (2011) Ustekinumab, a human interleukin-12/23 monoclonal antibody, in patients with psoriasis: a meta-analysis. J Dermatolog Treat 22: 323-336.
- Gordon KB, Leonardi CL, Lebwohl M, Blauvelt A, Cameron GS, et al. (2014) A 52-week, open-label study of the efficacy and safety of ixekizumab, an anti-interleukin-17A monoclonal antibody, in patients with chronic plaque psoriasis. J Am Acad Dermatol 71: 1176-1182.
- Mease PJ (2015) Inhibition of interleukin-17, interleukin-23 and the TH17 cell pathway in the treatment of psoriatic arthritis and psoriasis. Curr Opin Rheumatol 27: 127-133.
- Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, et al. (2007) Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol 8: 639-646.
- Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, et al. (2008) Human interleukin-17-producing cells originate from a CD161+ CD4+ T cell precursor. J Exp Med 205: 1903-1916.
- Maggi L, Santarlasci V, Capone M, Peired A, Frosali F, et al. (2010) CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. Eur J Immunol 40: 2174-2181.
- Miossec P, Kolls JK (2012) Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov 11: 763-776.
- Zhu S, Qian Y (2012) IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci (Lond) 122: 487-511.
- Cosmi L, Liotta F, Maggi E, Romagnani S, Annunziato F (2014) Th17 and non-classic Th1 cells in chronic inflammatory disorders: two sides of the same coin. Int Arch Allergy Immunol 164: 171-177.
- Chan JR, Blumenschein W, Murphy E, Diveu C, Wiekowski M, et al.(2006) IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J Exp Med 203: 2577-2587.
- Zhu S, Qian Y (2012) IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci (Lond) 122: 487-511.
- Hsu HC, Yang P, Wang J, Wu Q, Myers R, et al. (2008) Interleukin 17-producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol 9: 166-175.
- Rangel-Moreno J, Carragher DM, Luz Garcia-Hernandez M, Hwang JY, Kusser K, et al. (2011) The development of inducible bronchus-associated lymphoid tissue depends on IL-17. Nature Immunology 12: 639-646.
- Rogers PR, Dubey C, Swain SL (2000) Qualitative changes accompany memory T cell generation: faster, more effective responses at lower doses of antigen. JImmunol 164: 2338-2346.
- Sallusto F, Geginat J, Lanzavecchia A (2004) Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol 22: 745-763.
- Clark RA (2015) Resident memory T cells in human health and disease. Sci Transl Med 7: 269.
- Padovan E (2017) Modulation of CD4+ T Helper Cell Memory Responses in the Human Skin. Int Arch Allergy Immunol 173: 121-137.
- McLachlan JB, Catron DM, Moon JJ, Jenkins MK (2009) Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity 30: 277-288.
- Elyaman W, Kivis?¤kk P, Reddy J, Chitnis T, Raddassi K, et al. (2008) Distinct functions of autoreactive memory and effector CD4+ T cells in experimental autoimmune encephalomyelitis. Am J Pathol. 173: 411-422.
- Gaide O (2016) [Skin memory: the clinical implications]. Rev Med Suisse 12: 631-634.
- Mueller SN, Mackay LK (2016) Tissue-resident memory T cells: Local specialists in immune defence. Nat Rev Immunol 16: 79-89.
- Zaid A, Hor JL, Christo SN, Groom JR, Heath WR, et al. (2017) Chemokine Receptor-Dependent Control of Skin Tissue-Resident Memory T Cell Formation. J Immunol 199: 2451-2459.
- Pan Y, Kupper TS (2018) Metabolic Reprogramming and Longevity of Tissue-Resident Memory T Cells. Front Immunol 9: 1347.
- Cheuk S, Wikén M, Blomqvist L, Nylén S, Talme T, et al. (2014) Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 192: 3111-3120.
- Sérézal IG, Classon C, Cheuk S, Barrientos-Somarribas M, Wadman E, et al. (2018) Resident T Cells in Resolved Psoriasis Steer Tissue Responses that Stratify Clinical Outcome. J Invest Dermatol 138: 1754-1763.
- Kurihara K, Fujiyama T, Phadungsaksawasdi P, Ito T, Tokura Y (2019) Significance of IL-17A-producing CD8 + CD103 + skin resident memory T cells in psoriasis lesion and their possible relationship to clinical course. J Dermatol Sci 95: 21-27.
- Bhushan M, Bleiker TO, Ballsdon AE, Allen MH, Sopwith M, et al. (2002) Anti-E-selectin is ineffective in the treatment of psoriasis: A randomized trial. Br J Dermatol 146: 824-831.
- Boyman O, Hefti HP, Conrad C, Nickoloff BJ, Suter M, et al. (2004) Spontaneous development of psoriasis in a new animal model shows an essential role for resident T cells and tumor necrosis factor-alpha. J Exp Med 199: 731-736.
- Khairutdinov VR, Mikhailichenko AF, Belousova IE, Kuligina ES, Samtsov AV, et al. (2017) The role of intradermal proliferation of T-cells in the pathogenesis of psoriasis. An Bras Dermatol 92: 41-44.
- Tarcha EJ, Olsen CM, Probst P, Peckham D, Muñoz-Elías EJ, et al. (2017) Safety and pharmacodynamics of dalazatide, a Kv1.3 channel inhibitor, in the treatment of plaque psoriasis: A randomized phase 1b trial. PLoS One 12: 0180762.
- Bonefeld CM, Geisler C (2016) The role of innate lymphoid cells in healthy and inflamed skin. Immunol Lett 179: 25-28.
- Xiong T, Turner J-E (2018) Innate lymphoid cells in autoimmunity and chronic inflammatory diseases. Semin Immunopathol 40: 393-406.
- Walker JA, Barlow JL, McKenzie AN (2013) Innate lymphoid cells--how did we miss them? Nat Rev Immunol 13: 75-87.
- Teunissen MBM, Munneke JM, Bernink JH, Spuls PI, Res PCM, et al. (2014) Composition of Innate Lymphoid Cell Subsets in the Human Skin: Enrichment of NCR(+) ILC3 in Lesional Skin and Blood of Psoriasis Patients. J Invest Dermatol 134: 2351-2360.
- Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, et al. (2013) Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol 13: 145-149.
- Brüggen MC, Bauer WM, Reininger B, Clim E, Captarencu C, et al. (2016) In Situ Mapping of Innate Lymphoid Cells in Human Skin: Evidence for Remarkable Differences between Normal and Inflamed Skin. J Invest Dermatol 136: 2396-2405.
- Quaresma JAS (2019) Organization of the Skin Immune System and Compartmentalized Immune Responses in Infectious Diseases. Clin Microbiol Rev 32: 00034-00018.
- Pantelyushin S, Haak S, Ingold B, Kulig P, Heppner FL, et al. (2012) Rorγt+ innate lymphocytes and γδ T cells initiate psoriasiform plaque formation in mice. J Clin Invest 122: 2252-2256.
- Dyring-Andersen B, Geisler C, Agerbeck C, Lauritsen JP, Gúdjonsdottir SD et al. (2014) Increased number and frequency of group 3 innate lymphoid cells in nonlesional psoriatic skin. Br J Dermatol 170: 609-616.
- Villanova F, Flutter B, Tosi I, Grys K, Sreeneebus H, et al. (2014) Characterization of innate lymphoid cells in human skin and blood demonstrates increase of NKp44+ ILC3 in psoriasis. J Invest Dermatol 134: 984-991.
- Keren A, Shemer A, Ginzburg A, Ullmann Y, Schrum AG, et al. (2018) Innate lymphoid cells 3 induce psoriasis in xenotransplanted healthy human skin. J Allergy Clin Immunol 142: 305-308.
- Albanesi C, Scarponi C, Pallotta S, Daniele R, Bosisio D, et al. (2009) Chemerin expression marks early psoriatic skin lesions and correlates with plasmacytoid dendritic cell recruitment. J Exp Med 206: 249-258.
- Berthier-Vergnes O, Bermond F, Flacher V, Massacrier C, Schmitt D, et al. (2005) TNF-alpha enhances phenotypic and functional maturation of human epidermal Langerhans cells and induces IL-12 p40 and IP-10/CXCL-10 production. FEBS Lett 579: 3660-3668.
- Krueger JG, Bowcock A (2005) Psoriasis pathophysiology: Current concepts of pathogenesis. Ann Rheum Dis 64: 230-236.
- Chong SZ, Wong KL, Lin G, Yang CM, Wong SC, et al. (2011) Human CD8+ T cells drive Th1 responses through the differentiation of TNF/iNOS-producing dendritic cells. Eur J Immunol 41: 1639-16351.
- Wilsmann-Theis D, Koch S, Mindnich C, Bonness S, Schnautz S, et al. (2013) Generation and functional analysis of human TNF-α/iNOS-producing dendritic cells (Tip-DC). Allergy 68: 890-898.
- Lowes MA, Chamian F, Abello MV, Fuentes-Duculan J, Lin SL, et al. (2005) Increase in TNF-alpha and inducible nitric oxide synthase-expressing dendritic cells in psoriasis and reduction with efalizumab (anti-CD11a). Proc Natl Acad Sci USA 102: 19057-19062.
- Wang CQF, Akalu YT, Suarez-Farinas M, Gonzalez J, Mitsui H, et al. (2013) IL-17 and TNF synergistically modulate cytokine expression while suppressing melanogenesis: Potential relevance to psoriasis. J Invest Dermatol 133: 2741-2752.
- Valdimarsson H, Thorleifsdottir RH, Sigurdardottir SL, Gudjonsson JE, Johnston A (2009) Psoriasis--as an autoimmune disease caused by molecular mimicry. Trends Immunol 30: 494-501.
- Thorleifsdottir RH, Sigurdardottir SL, Sigurgeirsson B, Olafsson JH, Sigurdsson MI, et al. (2012) Improvement of psoriasis after tonsillectomy is associated with a decrease in the frequency of circulating T cells that recognize streptococcal determinants and homologous skin determinants. J Immunol 188: 5160-5165.
- Gudmundsdottir AS, Sigmundsdottir H, Sigurgeirsson B, Good MF, Valdimarsson H, et al. (1999) Is an epitope on keratin 17 a major target for autoreactive T lymphocytes in psoriasis? Clin Exp Immunol 117: 580-586.
- Baker BS, Laman JD, Powles A, van der Fits L, Voerman JS, et al. (2006) Peptidoglycan and peptidoglycan-specific Th1 cells in psoriatic skin lesions. J Pathol 209: 174-181.
- Qian L, Chen W, Sun W, Li M, Zheng R, et al. (2015) Antimicrobial peptide LL-37 along with peptidoglycan drive monocyte polarization toward CD14highCD16+ subset and may play a crucial role in the pathogenesis of psoriasis guttata. Am J Transl Res 7: 1081-1094.
- Baker BS, Powles A, Fry L (2006) Peptidoglycan: A major aetiological factor for psoriasis? Trends Immunol 27: 545-551.
- Flendrie M, Vissers WH, Creemers MC, de Jong EM, van de Kerkhof PC, et al. (2005) Dermatological conditions during TNF-alpha-blocking therapy in patients with rheumatoid arthritis: A prospective study. Arthritis Res Ther 7: 666-676.
- Palucka AK, Blanck JP, Bennett L, Pascual V, Banchereau J (2005) Cross-regulation of TNF and IFN-alpha in autoimmune diseases. Proc Natl Acad Sci USA 102: 3372-3377.
- Seneschal J, Milpied B, Vergier B, Lepreux S, Schaeverbeke T, et al. (2009) Cytokine imbalance with increased production of interferon-alpha in psoriasiform eruptions associated with antitumour necrosis factor-alpha treatments. Br J Dermatol 161: 1081-1088.
- Chen X, Bäumel M, Männel DN, Howard OM, Oppenheim JJ (2007) Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J Immunol 179: 154-161.
- Ma HL, Napierata L, Stedman N, Benoit S, Collins M, et al. (2010) Tumor necrosis factor alpha blockade exacerbates murine psoriasis-like disease by enhancing Th17 function and decreasing expansion of Treg cells. Arthritis Rheum 62: 430-440.
- Collamer AN, Guerrero KT, Henning JS, Battafarano DF (2008) Psoriatic skin lesions induced by tumor necrosis factor antagonist therapy: A literature review and potential mechanisms of action. Arthritis Rheum 59: 996-1001.
Citation: Patrushev AV, Zhukov AS, Khairutdinov VR, Samtsov AV (2022) Psoriasis Immunopathogenesis. J Clin Dermatol Ther 8: 096.
Copyright: © 2022 Patrushev A V, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.