RalA GTPase and MAP4K4 Function through NDR1 Activation in Stress Response and Apoptotic Signaling
*Corresponding Author(s):
Rasim SelimogluAnalysis Of Transduction Pathways Atp Group, Institut Curie, Paris, France
Tel:+330156246654,
Fax:+330156246650
Email:JCamonis.Institut.Curie@gmail.com
Audrey Bettoun
Analysis Of Transduction Pathways Atp Group, Institut Curie, Paris, France
Tel:+4402076790723,
Fax:+4402076796817
Email:a.hergovich@ucl.ac.uk
Abstract
The Ral GTPases are mainly known for their positive contribution to Ras-driven oncogenesis. In particular, it has been reported that RalA supports anchorage independent growth and cell transformation downstream of oncogenic Ras. However, one report describes RalA also as suppressor of early stages of Ras-induced carcinoma progression, proposing that RalA might also have tumor suppressive activities in specific settings, such as stress signaling. In tumorigenesis, regulation of Stress-Activated map Kinase (SAPK) pathways may determine cell survival or death in response to tumor environmental cues. While RalA regulates TNF-α signaling by contributing to SAPK activation, the role of Ral GTPases in response to other tumor environment driven stresses is largely unknown. Here, we describe the serine/threonine protein kinase NDR1 as a new partner of RalA signaling in control of SAPK. We report that under osmotic and oxidative stresses the Ste20-like MAP4K4 kinase, an effector of RalA via the Exocyst complex, directly phosphorylates NDR1 on Thr444, a key regulatory residue for NDR1 activation. Moreover, we found that apoptosis induction triggered by TNF-α cytokine treatment or RASSF1A over expression signals through the RalA-MAP4K4-NDR1 pathway. This novel and unexpected pro-apoptotic role of RalA suggests that the RalA GTPase can positively signal in tumor suppressor pathways, in addition to its proto-oncogenic role downstream of Ras.
Keywords
INTRODUCTION
The Ral GTPases are key actors in Ras-dependent oncogenesis [1]. The mammalian RalA and RalB GTPases play roles on different levels of tumorigenesis ranging from basic hallmarks of cancer, such as anchorage independent growth, to invasion and metastasis formation [2]. Ral GTPases belong to the Ras superfamily of small GTPases [3]. They are activated by Guanosine Exchange Factors (Ral GEFs), and are direct effectors of Ras in human tumorigenesis [4-7]. RalA functions in human cell transformation [4,8,9], while RalB promotes tumor cell survival and regulates cell motility [10,11]. More specifically, RalA loss-of-function inhibits anchorage independent growth of transformed human cells, and RalB knockdown causes tumor cell apoptosis and blocks cancer cell motility [2]. The specificity of the distinct biological functions of RalA and RalB has remained elusive, although different post-translational modifications of Ral which regulate Ral subcellular localization and activity are likely involved to separate functions [8,12-15]. However, this functional specificity of RalA and RalB is lost in mice, since knockout of both Ral GTPases is necessary to inhibit Ras-induced tumorigenesis [16], suggesting that RalA and RalB have redundant functions in this model system. Moreover, a recent report describes RalA as a suppressor of early stages of Ras-induced carcinoma progression [17], indicating that RalA might have tumor suppressive functions besides its reported oncogenic role downstream of Ras.
Downstream effectors of Ral GTPases include components of the Exocyst complex, Sec5 and Exo84, which support Ral functions in tumorigenesis [18]. It has been reported that the RalB-Sec5 complex activates TBK1 to promote tumor cell survival [10], and the RalB-Exo84 complex functions in autophagy [19]. However, the RalA-Exocyst signaling pathway remains poorly understood in cancer cells [20]. In this context of RalA-Exocyst signaling, the relationship of RalA to MAP4K4 is also yet to be explored in much more detail. MAP4K4 is a serine/threonine protein kinase of the GCK-IV family [21,22], and has been reported to support cellular transformation, tumor cell adhesion and invasion [23-25]. MAP4K4 is characterized by a unique C-terminal Citron Homology (CNH) domain [21], which is important for homodimerization and protein-protein interaction with Sec5, MEKK1 and beta1-integrin [10,26-28]. MAP4K4 has been defined as a Stress-Activated MAP Kinase (SAPK), driving the activation of MAPK-JNK cascades [22,29,30]. In Drosophila Msn, the fly counterpart of MAP4K4 [28], is part of a Ral-Sec5-Msn-JNK cascade regulating developmental-induced apoptosis [26]. However, the role of MAP4K4 as signaling partner of RalA and/or RalB has not been defined yet in human cells.
RalA mediates TNF-α-induced p38 MAPK activation and JNK-dependent activation of FOXO [26,31]. Moreover, the SAPK cascade can influence the fate of cancer cells in response to environmental and cellular stresses [32]. Together these findings suggested that RalA might regulate SAPK activation and stress responses. Here, we addressed these possible connections between RalA and MAP4K4 experimentally. More specifically, our study expands our understanding of how RalA signaling pathways mediate SAPK activation and determines the role of RalA in apoptosis induced responses to different types of environmental stresses. We identified MAP4K4 as a new upstream kinase of NDR1 (also known as STK38), an AGC serine/threonine protein kinase [33,34] that can be activated by Fas, TNF-α, osmotic and oxidative stresses [35-37]. Moreover, we demonstrate that RalA regulates NDR1 activation, via Exocyst and MAP4K4 signaling, to trigger apoptosis in response of extracellular stresses. Apoptosis triggered by overexpression of the tumor suppressor protein RASSF1A was also regulated by the RalA-Exocyst-MAP4K4-NDR1 pathway.
MATERIALS AND METHODS
Cell lines, reagents and plasmids
Yeast two-hybrid screens
Cell transfections
Cell stress experiments
In vitro phosphorylation of recombinant NDR1 by MAP4K4
NDR1 kinase assays
RESULTS
RalA, MAP4K4 and NDR1 mediate stress-activated p38 kinase phosphorylation
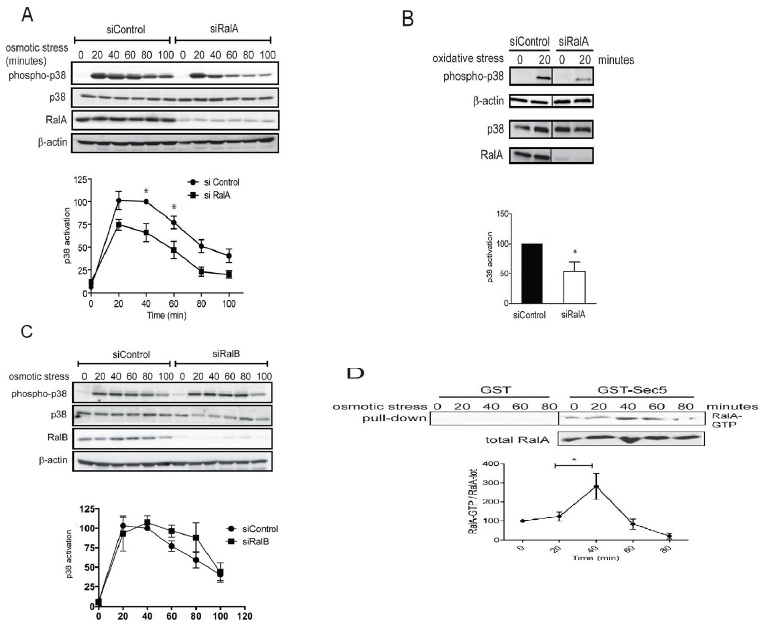
B. HeLa cells transfected with indicated siRNAs were stressed for 20 minutes with 400 μM H2O2 (oxidative stress) and processed for immunoblotting with indicated antibodies (Student t-test one tail, *P<0.05, n=3).
C. HeLa cells were transfected with indicated siRNAs and three days later 0.3 M sorbitol was added for indicated times. Cell lysates were subjected to immunoblotting using indicated antibodies. p38 activation kinetics upon osmotic stress were examined as described in A. Activation curves did not display statistically significant differences upon RalB depletion.
D. To determine RalA activation upon osmotic stress, HeLa cells were stressed with 0.3 M sorbitol as indicated and cell lysates were subjected to Ral-GTP pull-downs, followed by quantification of immunoblots. The increase of RalA-GTP levels at 40 minutes is significant (Student t-test two-tailed, **P<0.05, n=3). Non-specific binding to GST alone is also shown and was subtracted from the specific RalA-GTP pull downs to determine RalA-GTP levels.
In order to decipher the downstream molecular partners of RalA in stress signaling, we next studied the potential involvement of MAP4K4 (Figure 2A), a member of the GCK-IV kinase family and known effector of Ral signaling via the Exocyst complex in flies [26]. A role for MAP4K4 in p38/MAPK activation has already been reported previously in gastrulation and LPS-induced inflammation of macrophages [29,30], while the involvement of MAP4K4 in stress-induced p38 activation has not been explored yet. As observed in RalA-depleted cells (Figure 1A), knock down of MAP4K4 caused a 50% decrease of p38 activation upon osmotic stress induction (Figure 2A), suggesting that the MAP4K4-p38 pathway is triggered by osmotic stress downstream of RalA activation. To further elucidate the RalA-MAP4K4-p38 pathway, we conducted Yeast Two-Hybrid (Y2H) screens to identify novel players in this signaling cascade. Significantly, these screens revealed that Misshapen (Msn), the fly orthologue of human MAP4K4 [28], used as bait strongly interacted with the C-terminal hydrophobic motif of Tricornered (Trc) [50], the fly counterpart of human NDR1 [34] (Figure 2B). Reciprocally, a Y2H screen with full length human NDR1 as bait identified the CNH domain of MAP4K4 as interacting partner of NDR1 (Figure 2B). These Y2H data demonstrated that the interaction between MAP4K4 and NDR1 is conserved between flies and humans.
NDR1 belongs to the NDR/LATS subgroup of the AGC serine/threonine kinase family [33,34] and is activated by Fas, TNF-α, osmotic and oxidative stresses [35-37]. Therefore, our Y2H results suggested that NDR1 might function together with RalA and MAP4K4 upstream of p38 activation in the osmotic stress response. Supporting this hypothesis, NDR1 depletion decreased p38 activation under osmotic stress (Figure 2C). Two independent siRNAs specifically targeting NDR1 yielded similar results (Figure 2C and data not shown), showing that the observed effect is due to specific depletion of NDR1. NDR1 knockdown resulted in a similar p38 activation defect as observed in RalA- or MAP4K4-depleted cells (compare Figures 1A, 2A and 2C), supporting the notion that NDR1 can function together with RalA and MAP4K4 in osmotic stress signaling. Using a specific anti-phospho Thr444 NDR antibody (anti-T444-P) only detecting active NDR1 kinase phosphorylated at the key regulatory Thr444 residue in the hydrophobic motif [39-42], we further found that after 40 minutes of osmotic stress NDR1 was highly phosphorylated on Thr444 (Figure 2D), suggesting that NDR1 was activated upon osmotic stress induction. Taken together, these results demonstrate that RalA, MAP4K4 and NDR1 regulate p38 MAPK activation with similar kinetics and suggest that RalA, MAP4K4 and NDR1 may function in the same osmotic stress signaling pathway.
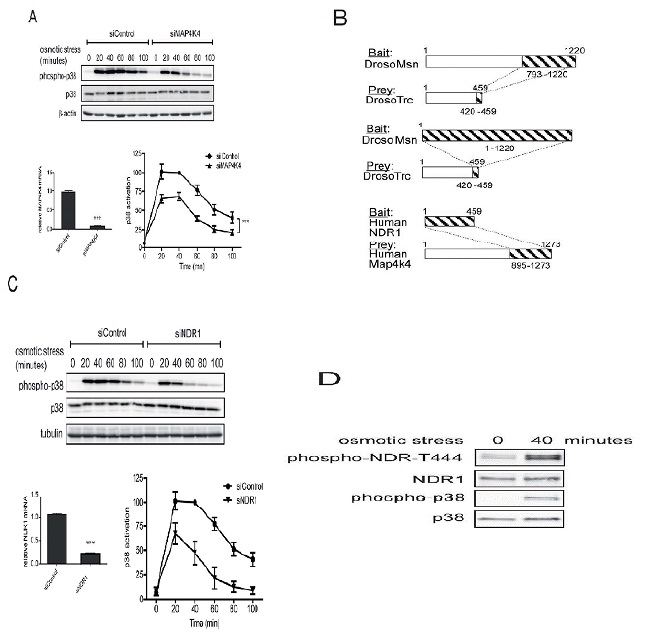
A. HeLa cells were transfected with indicated siRNAs and three days later 0.3 M sorbitol was added for indicated times. Cell lysates were subjected to SDS-PAGE and immunoblotted using indicated antibodies. p38 activation kinetics upon osmotic stress were quantified by comparing the ratios of phosphorylated (T180/Y182) with total p38. All values were normalized to the time point of 40 minutes of the siControl which was set as 100%. Activation curves displayed statistically significant differences (ANOVA, siControl vs. siMAP4K4, ***P<0.0002, n=3). The 20, 40 and 60 minutes time points were statistical different (Bonferroni test, siControl vs. siRalA P<0.05, n=3). MAP4K4 depletion was examined by mRNA quantification by real-time qPCR (siControl vs. siMAP4K4, ***P<0.0001, n=3).
B. Full-length Drosophila Misshapen (Msn) or its CNH domain (aa 793-1220) were used as baits to screen a Drosophila embryo library by Yeast Two-Hybrid (Y2H), revealing that the C-terminal region (aa 420-459) of Tricornered (Trc) interacted with Msn. Conversely, in a screen conducted on a human fetal brain library, full length human NDR1 kinase interacted with the CNH domain of MAP4K4 (aa 895-1273 of isoform 2). NDR1 and MAP4K4 are the human counterparts of Trc and Msn, respectively.
C. HeLa cells were transfected with indicated siRNAs and three days later 0.3 M sorbitol was added for indicated times. Cell lysates were subjected to immunoblotting using indicated antibodies. p38 activation kinetics upon osmotic stress were examined as described in A. Activation curves displayed statistically significant differences (ANOVA, siControl vs. siNDR1, ***P<0.0001, n=3). The 20, 40, 60 and 80 minutes time points were statistical different (Bonferroni test, siControl vs. siRalA P<0.05, n=3). NDR1 depletion was examined by mRNA quantification (siControl vs. siNDR1, P<0.0001, n=3).
D. Indicated HeLa cell lysates without or with osmotic stress (40 minutes with 0.3 M sorbitol) were subjected to Western blot analysis as indicated. In the phospho-NDR T444 blot the two bands correspond to the two isoforms of NDR kinases, NDR1 (lower band) and NDR2 (higher band).
The Ral GTPase effector MAP4K4 regulates NDR1 kinase by hydrophobic motif phosphorylation under stress.
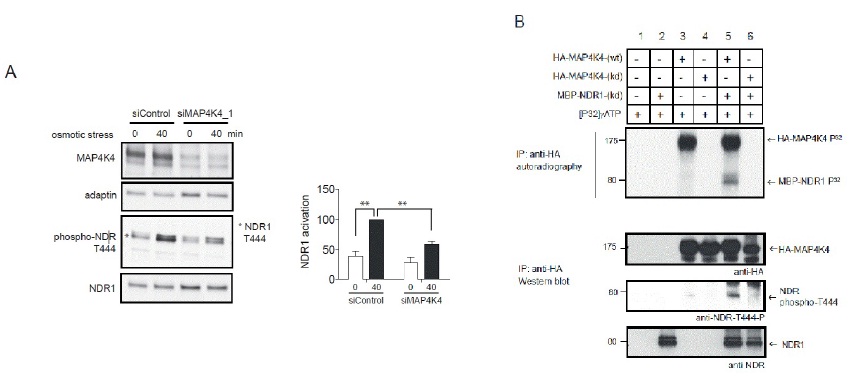
B. OA-treated lysates of Cos7 cells transiently expressing empty vector (lanes 1 and 2), wild-type HA-MAP4K4 (wt; lanes 3 and 5) or kinase-dead HA-MAP4K4 (kd; lanes 4 and 6) were subjected to immunoprecipitation with anti-HA 12CA5 antibody. Immuno-purified proteins were then processed for standard kinase assays without (lanes 1, 3 and 4) or with MBP-NDR1 kinase-dead (kd) (lanes 2, 5 and 6), followed by SDS-PAGE and autoradiography (upper panel). The positions of HA-MAP4K4 and MBP-NDR1 (kd) are indicated by arrows. K, relative molecular mass in thousands. In lower panels, identical kinase assays were performed using unlabelled ATP and MBP-NDR1 (kd) as a substrate followed by immunoblotting using indicated antibodies.
RalA and Exocyst regulate NDR1 activation upon several stresses
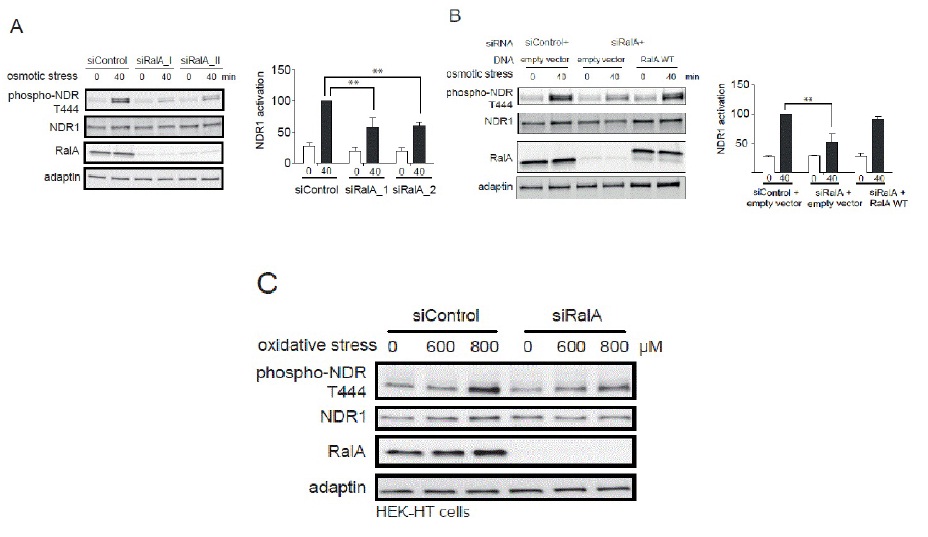
B. HeLa cells were first transfected with indicated siRNAs, followed by transfection with indicated plasmids (empty vector, plasmid without insert; RalA WT, siRNA resistant myc-RalA). Cell lysates were analyzed as described above in section A (Student t-test, **P<0.05, n=3). Noteworthy, the effect caused by RalA depletion was suppresses by the expression of siRNA-resistant RalA.
C. HEK-HT cells transfected with indicated siRNAs were stressed with 600 or 800 μM H2O2 and processed for immunoblotting using indicated antibodies.
The importance of RalA in NDR1 kinase activation under stress was further evaluated by measuring NDR1 kinase activity using a specific NDR substrate [44]. In control cells, immunoprecipitated endogenous NDR1 from cells treated by 0.3 M sorbitol was able to phosphorylate the GST-PHO1 peptide containing the NDR kinase substrate signature (Figure 5A). This phosphorylation was decreased upon depletion of NDR1 (Figure 5B) or absent when a scrambled peptide substrate was used (data not shown). Immunoprecipitated NDR1 from RalA-depleted cells showed a decreased capacity in peptide phosphorylation in contrast to controls (Figure 5A). These results demonstrate that RalA is necessary for normal NDR1 kinase activation under osmotic stress, hence establishing RalA as novel upstream regulator of NDR1 in osmotic stress response signaling.
The Sec5 subunit of the octameric Exocyst complex constitutes a molecular link between MAP4K4 and Ral in flies [26]. Within the Exocyst complex, two subunits, Sec5 and Exo84 interact with RalA and RalB [53,54] and execute Ral-dependent signal transduction [10,12,19,43,55]. Consequently, the involvement of Sec5 and Exo84 in RalA and MAP4K4-dependent NDR1 phosphorylation was tested (Figure 5C). Sec5 or Exo84 were independently depleted in HeLa cells followed by osmotic stress treatment. As observed in RalA- or MAP4K4-depleted cells, knock-down of Sec5 or Exo84 alone abolished NDR1 phosphorylation upon osmotic stress (Figure 5C), indicating that the Exocyst complex is also functioning upstream of NDR1 in osmotic stress signaling.
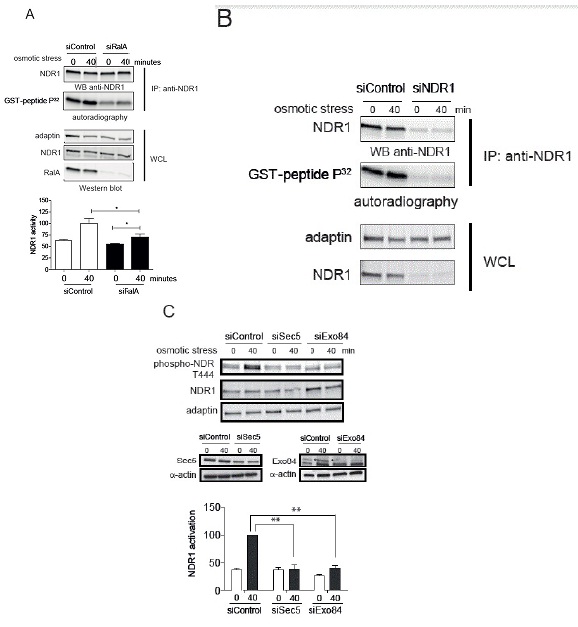
B. HeLa cells were transfected with indicated siRNAs, followed by stimulation for 40 minutes with 0.3 M sorbitol. Endogenous NDR1 kinase activity was analyzed as described in panel A.
C. HeLa cells were transfected with indicated siRNAs, treated with 0.3 M sorbitol for indicated times, followed by processing of cell lysates for immunoblotting with indicated antibodies. Compared to siControl, Sec5 or Exo84 were depleted at 70% in cells. The specific signal for Exo84 is indicated by an asterisk. NDR1 activation was examined as described in Figure 3A. Of note, depletion of Sec5 or Exo84 significantly inhibited NDR1 activation (Student t-test two-tailed, **P<0.05, n=3).
RalA and MAP4K4 are upstream regulators of NDR1 in stress-induced apoptosis signaling
As reported [36] upon TNF-α treatment, NDR1 phosphorylation was induced after 1 hour of treatment and cleaved caspase-3 appeared after 3 hours (Figure 6B). RalA depletion caused a decrease of 50% in both NDR1 and caspase 3 activation (Figure 6B). In contrast, despite the reported role of RalB in keeping apoptosis at bay to promote cancer cell survival [11], TNF-α activated caspase 3 and NDR1 in RalB-depleted cells similarly to control cells (data not shown), suggesting that RalB is not required for TNF-α-induced apoptosis in this setting. Furthermore, in MAP4K4-depleted cells, TNF-α induced caspase 3 cleavage and NDR1 phosphorylation were reduced (Figure 6C). In summary, these results indicate that RalA and MAP4K4 represent a crucial link between the activation of the pro-apoptotic signaling pathway and the onset of an NDR1-dependent apoptotic program triggered by TNF-α.
In the apoptosis pathway, NDR1 also functions downstream of the tumor suppressor protein RASSF1A [36]. In HeLa cells, RASSF1A overexpression promotes apoptosis by inducing NDR1 phosphorylation, which triggers a pro-apoptotic program, resulting in PARP cleavage [36]. In these conditions depletion of RalA caused a decrease in RASSF1A overexpression-induced NDR1 phosphorylation and PARP cleavage by 50% (Figure 6D). In support of our notion that RalA and MAP4K4 act upstream of NDR1 in apoptosis induction, silencing of MAP4K4 by two independent siRNAs also decreased NDR1 phosphorylation and accumulation of cleaved PARP upon apoptosis induction by RASSF1A over expression (Figure 6E). The level of MAP4K4 depletion correlated with decreased NDR1 activation and PARP cleavage (Figure 6E). Taken together, these results show that the RalA-MAP4K4-NDR1 pathway plays a role in the TNF-α- and RASSF1A-driven apoptotic responses.
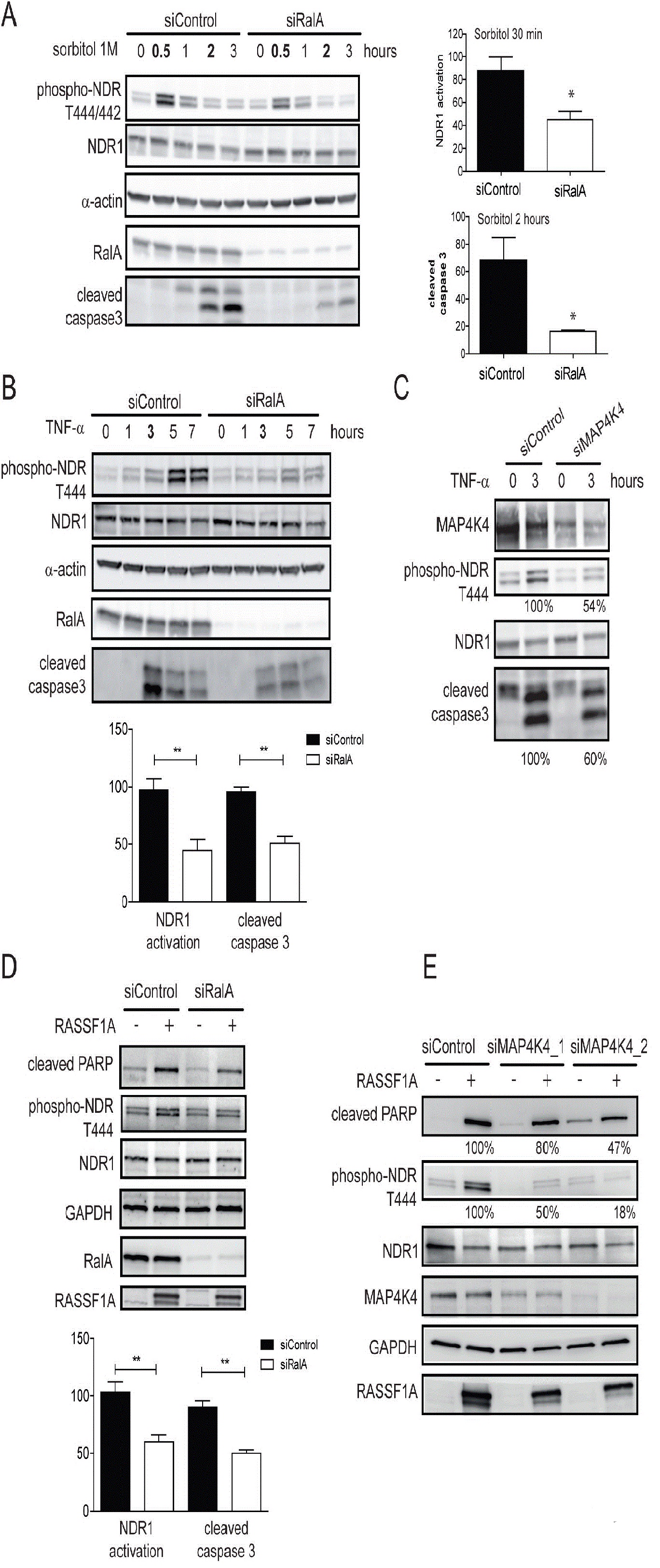
The participation of RalA in SAPK pathways in response to TNF- α and oxidative stress signaling has been previously described [26,31]. However, we define here for the first time the signaling pathway function downstream of RalA in response to these stresses. Our results reveal a new RalA-Exocyst-MAP4K4-NDR1 pathway in control of osmotic and oxidative stress signaling. Mechanistically, our data suggest that RalA through the Exocyst complex signals to the Ste20-like MAP4K4 kinase, which in turn activates the NDR1 kinase by phosphorylating the hydrophobic motif of NDR1. Consequently, our data show that RalA and MAP4K4 are upstream regulators of NDR1 in osmotic stress-, TNF-α -and RASSF1A-induced apoptotic programs.
DISCUSSION
Deconvolution of the means and pathways used by Ras oncogenes has largely been a result of the disentangling of the Ras interactome into three main branches: the Raf-MEK-ERK kinase cascade, the PI3K-AKT kinase pathway, and equally important the Ral-dependent branch [20]. Within the Ral-dependent branch human alA and RalB are essential and in some context instructive for oncogenic transformation [2]. In regard of the oncogenic aspect, RalA and RalB play different biochemical and biological roles, although both, RalA and RalB, utilize the same signal transduction platform, namely the Exocyst complex [2]. Our understanding of RalB signaling and functions has steadily progressed by discoveries showing that RalB plays a role in the capacity of cancer cells to dodge apoptosis [11], and by elucidating RalB’s permissive and instructive role in autophagy [19]. In contrast, RalA beyond the Exocyst complex has found less solid grounds to justify its apparently contradictory functions in cellular transformation [20]. On the one hand, current evidence suggests that RalA is needed for tumorigenesis by playing a positive role in anchorage independent growth of cancer cells [4,8,9] and by also regulating the turnover of cadherins at their physiological plasma membrane localization [58]. On the other hand, RalA paradoxically can display tumor suppressive properties in squamous cell carcinoma progression [17].
iming at establishing a comprehensive RalA interactome, we undertook a systematic Y2H approach through which we uncovered and validated a novel RalA-Exocyst-MAP4K4-NDR1 signal transduction pathway that is necessary for efficient stress and apoptosis signaling in our settings (Figure 7). This discovery will not only fuel future studies of RalA-related cell biology, but also reveals an overlap between the RalA pathway and NDR-Hippo signaling, in which NDR1 functions as central player [33]. Our study also sheds light on a new signaling wiring upstream of the NDR1 kinase (Figure 7). More specifically, in addition to being regulated by the MST1/2 Hippo kinases [36,40] and MST3 [52,59], we show here that the NDR1 kinase can also be regulated by other members of the Ste20-like kinase family, namely the MAP4K4 kinase in our settings. This observation revealed that a Ste20-like kinase outside of the GCKII and GCKIII subfamilies of Ste20-like kinases is required for the activation of NDR1 by hydrophobic motif phosphorylation. This suggests an additional level of redundancy for NDR1 activation to ensure a proper regulation of the tumor suppressive NDR1 kinase [60]. Perhaps, given that the regulation of NDR1 by MST1 is dependent on the co-activator MOB1 [33,61], the regulation of NDR1 by MAP4K4 represents an hMOB1-independent level. Therefore, future research into the regulation of the RalA-Exocyst-MAP4K4-NDR1 pathway by the tumor suppressor MOB1 is warranted.
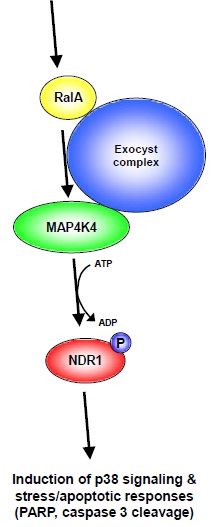
Figure 7: Model of RalA-Exocyst-MAP4K4-NDR1 stress/apoptotic signaling.
Based on our findings we are tempted to propose a model illustrating how RalA and NDR1 can function together in stress/apoptotic signaling. Upon activation of RalA by stress (e.g. osmotic or oxidative stress) and/or apoptotic stimuli, RalA supports the activation of MAP4K4 facilitated by the Exocyst complex. Once activated MAP4K4 phosphorylates and thereby activates NDR1 kinase, which subsequently modulates p38 signaling and the response to stress and/or apoptotic stimuli. Notably, our model does not exclude the possibility that each component can also be regulated by additional factors at each individual signaling level.
Our work shows further that the RalA-Exocyst-MAP4K4-NDR1 cascade can be activated by different stress stimuli, such as TNF-? which triggers apoptotic signaling or sorbitol which mimics high osmotic stress. Therefore, the RalA-Exocyst-MAP4K4-NDR1 pathway is likely to play a role in a broader stress signaling spectrum. Particularly, the pro-apoptotic role of NDR1 downstream of RalA-MAP4K4 signaling could be of specific interest in selected cancer settings. In this context, the pro-apoptotic role of NDR1 downstream of RalA-MAP4K4 signaling is in full agreement with the previously reported pro-apoptotic function of NDR1 in the prevention of T-cell lymphoma formation [60]. Therefore, future clinical and animal studies addressing the pro-apoptotic role of RalA-MAP4K4-NDR1 signaling in complex multicellular contexts are warranted.
Given that we also observed that the RalA-MAP4K4-NDR1 axis is required for apoptotic signaling upon RASSF1A overexpression, this pro-apoptotic role of RalA highlights a potential paradoxical role of RalA in tumor suppression vs. tumor promotion. In this regard, it is worth mentioning that it has been already documented that at early stages of cancer progression RalA suppression is required for Ras-dependent invasive properties of squamous carcinoma cells associated with E-cadherin loss [17]. Therefore, current evidence supports paradoxically opposite roles for RalA in tumor progression and suppression, which appear to be contradictory. On the one hand, RalA supports cellular transformation downstream of Ras [4,8,9]. On the other hand, our work reported herein suggests that RalA might also function in tumor suppressor signaling by promoting cancer cell apoptosis. Our findings further propose that the RalA GTPase could control tumor environment-induced cell death, while RalB plays a role in the cell autonomous survival of cancer cells [10,11]. Therefore, future research addressing this paradox is warranted to elucidate the mechanistic switch dictating whether RalA should support tumor promotion or tumor suppression. Nevertheless, our work provides the first molecular lead into how RalA could execute a role in tumor suppression. Based on our findings it is tempting to speculate that at first RalA would support stress-related signaling pathways, which in case of excessive stress would interfere with tumor growth. Thus, our work opens new perspectives into the contribution of RalA to tumor environment stress sensing.
ACKNOWLEDGMENT
We thank G. Scita (IEO, Milano, Italy) for providing reagents and all Hybrigenics staff for Y2H analysis. This work was supported by a GenHomme Network Grant (02490-6088) to Hybrigenics and Institut Curie, and by Fondation ARC pour la Recherche sur le Cancer , by Ligue National contre le Cancer to MCP (RS14/75-54), and association Christelle Bouillot RS was supported by ARC; AB and CJ were supported by Ligue Nationale contre le cancer, and CJ also by INCA, respectively. AH is a Wellcome Trust Research Career Development fellow (090090/Z/09/Z) at the UCL Cancer Institute. The work was also supported by the grant ANR-08-NLAN-0290-01 to J.C.
REFERENCES
- Feig LA (2003) Ral-GTPases: approaching their 15 minutes of fame. Trends Cell Biol 13: 419-425.
- Bodemann BO, White MA (2008) Ral GTPases and cancer: linchpin support of the tumorigenic platform. Nat Rev Cancer 8: 133-140.
- Chardin P, Tavitian A (1986) The ral gene: a new ras related gene isolated by the use of a synthetic probe. Embo J 5: 2203-2208.
- Lim KH, Baines AT, Fiordalisi JJ, Shipitsin M, Feig LA, et al. (2005) Activation of RalA is critical for Ras-induced tumorigenesis of human cells. Cancer Cell 7: 533-545.
- Martin TD, Samuel JC, Routh ED, Der CJ, Yeh JJ (2011) Activation and involvement of Ral GTPases in colorectal cancer. Cancer Res 71: 206-215.
- Vigil D, Martin TD, Williams F, Yeh JJ, Campbell SL, et al. (2010) Aberrant overexpression of the Rgl2 Ral small GTPase-specific guanine nucleotide exchange factor promotes pancreatic cancer growth through Ral-dependent and Ral-independent mechanisms. J Biol Chem 285: 34729-34740.
- Zipfel PA, Brady DC, Kashatus DF, Ancrile BD, Tyler DS, et al. (2010) Ral activation promotes melanomagenesis. Oncogene 29: 4859-4864.
- Lim KH, Brady DC, Kashatus DF, Ancrile BB, Der CJ, et al. (2010) Aurora-A phosphorylates, activates, and relocalizes the small GTPase RalA. Mol Cell Biol 30: 508-523.
- Lim KH, O'Hayer K, Adam SJ, Kendall SD, Campbell PM, et al. (2006) Divergent roles for RalA and RalB in malignant growth of human pancreatic carcinoma cells. Curr Biol 16: 2385-2394.
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, et al. (2006) RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell 127: 157-170.
- Chien Y, White MA (2003) RAL GTPases are linchpin modulators of human tumour-cell proliferation and survival. EMBO Rep 4: 800-806.
- Neyraud V, Aushev VN, Hatzoglou A, Meunier B, Cascone I, et al. (2012) RalA and RalB proteins are ubiquitinated GTPases, and ubiquitinated RalA increases lipid raft exposure at the plasma membrane. J Biol Chem 287: 29397-29405.
- Sablina AA, Chen W, Arroyo JD, Corral L, Hector M, et al. (2007) The tumor suppressor PP2A Abeta regulates the RalA GTPase. Cell 129: 969-982.
- Wang H, Owens C, Chandra N, Conaway MR, Brautigan DL, et al. (2010) Phosphorylation of RalB is important for bladder cancer cell growth and metastasis. Cancer Res 70: 8760-8769.
- Wu JC, Chen TY, Yu CT, Tsai SJ, Hsu JM, et al. (2005) Identification of V23RalA-Ser194 as a critical mediator for Aurora-A-induced cellular motility and transformation by small pool expression screening. J Biol Chem 280: 9013-9022.
- Peschard P, McCarthy A, Leblanc-Dominguez V, Yeo M, Guichard S, et al. (2012) Genetic deletion of RALA and RALB small GTPases reveals redundant functions in development and tumorigenesis. Curr Biol 22: 2063-2068.
- Sowalsky AG, Alt-Holland A, Shamis Y, Garlick JA, Feig LA (2010) RalA suppresses early stages of Ras-induced squamous cell carcinoma progression. Oncogene 29: 45-55.
- Camonis JH, White MA (2005) Ral GTPases: corrupting the exocyst in cancer cells. Trends Cell Biol 15: 327-332.
- Bodemann BO, Orvedahl A, Cheng T, Ram RR, Ou YH, et al. (2011) RalB and the exocyst mediate the cellular starvation response by direct activation of autophagosome assembly. Cell 144: 253-267.
- Stephen AG, Esposito D, Bagni RK, McCormick F (2014) Dragging ras back in the ring. Cancer Cell 25: 272-281.
- Dan I, Watanabe NM, Kusumi A (2001) The Ste20 group kinases as regulators of MAP kinase cascades. Trends Cell Biol 11: 220-230.
- Yao Z, Zhou G, Wang XS, Brown A, Diener K, et al. (1999) A novel human STE20-related protein kinase, HGK, that specifically activates the c-Jun N-terminal kinase signaling pathway. J Biol Chem 274: 2118-2125.
- Collins CS, Hong J, Sapinoso L, Zhou Y, Liu Z, et al. (2006) A small interfering RNA screen for modulators of tumor cell motility identifies MAP4K4 as a promigratory kinase. Proc Natl Acad Sci USA 103: 3775-3780.
- Liang JJ, Wang H, Rashid A, Tan TH, Hwang RF, et al. (2008) Expression of MAP4K4 is associated with worse prognosis in patients with stage II pancreatic ductal adenocarcinoma. Clin Cancer Res 14: 7043-7049.
- Wright JH, Wang X, Manning G, LaMere BJ, Le P, et al. (2003) The STE20 kinase HGK is broadly expressed in human tumor cells and can modulate cellular transformation, invasion, and adhesion. Mol Cell Biol 23: 2068-2082.
- Balakireva M, Rossé C, Langevin J, Chien YC, Gho M, et al. (2006) The Ral/exocyst effector complex counters c-Jun N-terminal kinase-dependent apoptosis in Drosophila melanogaster. Mol Cell Biol 26: 8953-8963.
- Poinat P, De Arcangelis A, Sookhareea S, Zhu X, Hedgecock EM, et al. (2002) A conserved interaction between beta1 integrin/PAT-3 and Nck-interacting kinase/MIG-15 that mediates commissural axon navigation in C. elegans. Curr Biol 12: 622-631.
- Su YC, Han J, Xu S, Cobb M, Skolnik EY (1997) NIK is a new Ste20-related kinase that binds NCK and MEKK1 and activates the SAPK/JNK cascade via a conserved regulatory domain. EMBO J 16: 1279-1290.
- Aouadi M, Tesz GJ, Nicoloro SM, Wang M, Chouinard M, et al. (2009) Orally delivered siRNA targeting macrophage Map4k4 suppresses systemic inflammation. Nature 458: 1180-1184.
- Zohn IE, Li Y, Skolnik EY, Anderson KV, Han J, et al. (2006) p38 and a p38-interacting protein are critical for downregulation of E-cadherin during mouse gastrulation. Cell 125: 957-969.
- Essers MA, Weijzen S, de Vries-Smits AM, Saarloos I, de Ruiter ND, et al. (2004) FOXO transcription factor activation by oxidative stress mediated by the small GTPase Ral and JNK. Embo J 23: 4802-4812.
- Coulthard LR, White DE, Jones DL, McDermott MF, Burchill SA (2009) p38(MAPK): stress responses from molecular mechanisms to therapeutics. Trends Mol Med 15: 369-379.
- Hergovich A (2013) Regulation and functions of mammalian LATS/NDR kinases: looking beyond canonical Hippo signalling. Cell Biosci 3: 32.
- Hergovich A, Stegert MR, Schmitz D, Hemmings BA (2006) NDR kinases regulate essential cell processes from yeast to humans. Nat Rev Mol Cell Biol 7: 253-264.
- Fuller SJ, Pikkarainen S, Tham el L, Cullingford TE, Molkentin JD, et al. (2008) Nuclear Dbf2-Related protein kinases (NDRs) in isolated cardiac myocytes and the myocardium: activation by cellular stresses and by phosphoprotein serine-/threonine-phosphatase inhibitors. Cell Signal 20: 1564-1577.
- Vichalkovski A, Gresko E, Cornils H, Hergovich A, Schmitz D, et al. (2008) NDR kinase is activated by RASSF1A/MST1 in response to Fas receptor stimulation and promotes apoptosis. Curr Biol 18: 1889-1895.
- Zhou Y, Adolfs Y, Pijnappel WW, Fuller SJ, Van der Schors RC, et al. (2011) MICAL-1 is a negative regulator of MST-NDR kinase signaling and apoptosis. Mol Cell Biol 31: 3603-3615.
- Hahn WC, Counter CM, Lundberg AS, Beijersbergen RL, Brooks MW, et al. (1999) Creation of human tumour cells with defined genetic elements. Nature 400: 464-468.
- Hergovich A, Bichsel SJ, Hemmings BA (2005) Human NDR kinases are rapidly activated by MOB proteins through recruitment to the plasma membrane and phosphorylation. Mol Cell Biol 25: 8259-8272.
- Hergovich A, Kohler RS, Schmitz D, Vichalkovski A, Cornils H, et al. (2009) The MST1 and hMOB1 tumor suppressors control human centrosome duplication by regulating NDR kinase phosphorylation. Curr Biol 19: 1692-1702.
- Hergovich A, Lamla S, Nigg EA, Hemmings BA (2007) Centrosome-associated NDR kinase regulates centrosome duplication. Mol Cell 25: 625-634.
- Tamaskovic R, Bichsel SJ, Rogniaux H, Stegert MR, Hemmings BA (2003) Mechanism of Ca2+-mediated regulation of NDR protein kinase through autophosphorylation and phosphorylation by an upstream kinase. J Biol Chem 278: 6710-6718.
- Cascone I, Selimoglu R, Ozdemir C, Del Nery E, Yeaman C, et al. (2008) Distinct roles of RalA and RalB in the progression of cytokinesis are supported by distinct RalGEFs. EMBO J 27: 2375-2387.
- Millward TA, Heizmann CW, Schäfer BW, Hemmings BA (1998) Calcium regulation of Ndr protein kinase mediated by S100 calcium-binding proteins. EMBO J 17: 5913-5922.
- Vojtek AB, Hollenberg SM (1995) Ras-Raf interaction: two-hybrid analysis. Methods Enzymol 255: 331-342.
- Bartel P, Chien CT, Sternglanz R, Fields S (1993) Elimination of false positives that arise in using the two-hybrid system. Biotechniques 14: 920-924.
- Fromont-Racine M, Rain JC, Legrain P (2002) Building protein-protein networks by two-hybrid mating strategy. Methods Enzymol 350: 513-524.
- Luo J, Solimini NL, Elledge SJ (2009) Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136: 823-837.
- Burg MB, Ferraris JD, Dmitrieva NI (2007) Cellular response to hyperosmotic stresses. Physiol Rev 87: 1441-1474.
- Geng W, He B, Wang M, Adler PN (2000) The tricornered gene, which is required for the integrity of epidermal cell extensions, encodes the Drosophila nuclear DBF2-related kinase. Genetics 156: 1817-1828.
- Millward TA, Hess D, Hemmings BA (1999) Ndr protein kinase is regulated by phosphorylation on two conserved sequence motifs. J Biol Chem 274: 33847-33850.
- Cornils H, Kohler RS, Hergovich A, Hemmings BA (2011) Human NDR kinases control G(1)/S cell cycle transition by directly regulating p21 stability. Mol Cell Biol 31: 1382-1395.
- Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, et al. (2002) The exocyst is a Ral effector complex. Nat Cell Biol 4: 66-72.
- Moskalenko S, Tong C, Rosse C, Mirey G, Formstecher E, et al. (2003) Ral GTPases regulate exocyst assembly through dual subunit interactions. J Biol Chem 278: 51743-51748.
- Parrini MC, Sadou-Dubourgnoux A, Aoki K, Kunida K, Biondini M, et al. (2011) SH3BP1, an exocyst-associated RhoGAP, inactivates Rac1 at the front to drive cell motility. Mol Cell 42: 650-661.
- Rosette C, Karin M (1996) Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 274: 1194-1197.
- Marfe G, Pucci B, De Martino L, Fiorito F, Di Stefano C, et al. (2009) Heat-shock pretreatment inhibits sorbitol-induced apoptosis in K562, U937 and HeLa cells. Int J Cancer 125: 2077-2085.
- Shipitsin M, Feig LA (2004) RalA but not RalB enhances polarized delivery of membrane proteins to the basolateral surface of epithelial cells. Mol Cell Biol 24: 5746-5756.
- Stegert MR, Hergovich A, Tamaskovic R, Bichsel SJ, Hemmings BA (2005) Regulation of NDR protein kinase by hydrophobic motif phosphorylation mediated by the mammalian Ste20-like kinase MST3. Mol Cell Biol 25: 11019-11029.
- Cornils H, Stegert MR, Hergovich A, Hynx D, Schmitz D, et al. (2010) Ablation of the kinase NDR1 predisposes mice to the development of T cell lymphoma. Sci Signal 3: ra47.
- Hergovich A (2011) MOB control: reviewing a conserved family of kinase regulators. Cell Signal 23: 1433-1440.
Citation: Mycielska ME, Wachsmuth CJ, Dettmer K, Wagner C, Schlitt HJ, et al. (2014) Hsp90 Inhibition Affects Cell Metabolism by Disrupting Mitochondrial Protein Insertion. J Cell Biol Cell Metab 1: 001.
Copyright: © 2014 Rasim Selimoglu, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.