Journal of Modern Chemical Sciences Category: Chemistry
Type: Research Article
Recovery of the Antibiotic Activity against Resistant Bacterial Strains by Selective Guanidinylation of the 3-Methylamino Group of Gentamicin
*Corresponding Author(s):
Julia RevueltaDepartmento De Quimica Bioorganica, Instituto De Química Orgánica General (CSIC) Juan De La Cierva, 3. 28006-Madrid, Spain
Tel:+34 912587487,
Email:andres.g.santana@csic.es
Andrés G Santana
Departmento De Quimica Bioorganica, Instituto De Química Orgánica General (CSIC) Juan De La Cierva, 3. 28006-Madrid, Spain
Tel:+34 91 2587455,
Email:andres.g.santana@csic.es
Received Date: Jul 11, 2017
Accepted Date: Sep 12, 2017
Published Date: Sep 26, 2017
Abstract
Aminoglycoside antibiotics are highly potent, wide-spectrum bactericidals. Unfortunately, their use in clinical practice has been seriously limited as a result of their toxicity and susceptibility to enzymatic inactivation. Herein, we describe the synthesis and biological properties of a new gentamicin derivative (C1a and C2 gentamicin components). The selective modification of N-3″ in this antibiotic not only maintains an antimicrobial activity similar to the parent aminoglycoside but also against resistant strains expressing several AMEs (AAC (6′)-Ib, APH(2″), ANT(6) and APH(3′)).
INTRODUCTION
Ever since their discovery in the mid XX century, aminoglycoside antibiotics have become one of the go to treatments for bacterial infections. The first congener of this family, streptomycin was isolated in 1944 and soon after, several members of this class of compounds were also reported [1]. From a chemical point of view, aminoglycosides are water soluble polycationic pseudo-oligosaccharides constituted by amino sugars attached to a cyclitol by glycosidic linkages. Their potent bactericidal activity relies on binding specifically to the decoding A site of RNA in the bacterial small ribosomal subunit, thus interfering with protein synthesis [2,3]. These compounds are widely used in the treatment of serious infections caused by gram negative/gram positive bacteria, although their clinical use is associated with dose-limiting nephrotoxicity and ototoxicity [4,5]. Worse yet, many of the aminoglycoside antibiotics originally identified are no longer clinically useful as they are compromised by bacterial resistance mechanisms [6].
Acquired resistance to aminoglycoside antibiotics can occur via three different mechanisms: Mutation of the ribosomal target, reduced permeability for the antibiotics, and enzymatic modification of the drugs, thus leading to inactivation. From a clinical point of view, the most relevant source of resistance is the enzymatic inactivation of the drugs by modification of its amine or hydroxyl groups. Antibiotic Modifying Enzymes (AMEs) can be broadly classified as N-Acetyltransferases (AACs), O-Adenyl Transferases (ANTs) and O-Phosphotransferases (APHs). Each family involves enzymes that catalyse the same reactions but with different regioselectivity and substrate specificity (Figure 1).
With this in mind, and given the urgent demand for the discovery of new antibiotics to overcome the resistance to these compounds, considerable effort has been devoted to designing new semisynthetic aminoglycoside antibiotics that are immune to inactivation [7]. The intrinsic potency of aminoglycosides makes them excellent candidates to explore new ways to avoid bacterial resistance and diminish toxicity. Our group has been actively involved in the design of new antibiotics that are not susceptible to inactivation by AMEs. With this purpose, different design strategies have been employed (Figure 1) [8].
These derivatives included changes in the distribution of the amino groups (Figure 1a-c) [9], conformational restriction of the drug (Figure 1d) [10], the simultaneous incorporation of kanamycin and ribostamycin fragments within the same antibiotic scaffold (Figure 1e) [11], aminoglycoside dimerisation (Figure 1f) [12] and the introduction of bulky substituent’s properly positioned to interfere with drug recognition (Figure 1g) [13]. Unfortunately, despite their structural diversity, these compounds are substrates of the AMEs to some extent and some of them even lost their antibiotic activity. More recently, we have demonstrated that the A site shows a clear tolerance for modification at the N-3″ position of the aminoglycosides [14,15] and that the guanidinylation of this position maintains the antibiotic activity against aminoglycoside 6′-acetyl-transferase and 4′-nucleotidyl-transfersase-expressing strains (Figure 1h) [16, 17].
Acquired resistance to aminoglycoside antibiotics can occur via three different mechanisms: Mutation of the ribosomal target, reduced permeability for the antibiotics, and enzymatic modification of the drugs, thus leading to inactivation. From a clinical point of view, the most relevant source of resistance is the enzymatic inactivation of the drugs by modification of its amine or hydroxyl groups. Antibiotic Modifying Enzymes (AMEs) can be broadly classified as N-Acetyltransferases (AACs), O-Adenyl Transferases (ANTs) and O-Phosphotransferases (APHs). Each family involves enzymes that catalyse the same reactions but with different regioselectivity and substrate specificity (Figure 1).
With this in mind, and given the urgent demand for the discovery of new antibiotics to overcome the resistance to these compounds, considerable effort has been devoted to designing new semisynthetic aminoglycoside antibiotics that are immune to inactivation [7]. The intrinsic potency of aminoglycosides makes them excellent candidates to explore new ways to avoid bacterial resistance and diminish toxicity. Our group has been actively involved in the design of new antibiotics that are not susceptible to inactivation by AMEs. With this purpose, different design strategies have been employed (Figure 1) [8].
These derivatives included changes in the distribution of the amino groups (Figure 1a-c) [9], conformational restriction of the drug (Figure 1d) [10], the simultaneous incorporation of kanamycin and ribostamycin fragments within the same antibiotic scaffold (Figure 1e) [11], aminoglycoside dimerisation (Figure 1f) [12] and the introduction of bulky substituent’s properly positioned to interfere with drug recognition (Figure 1g) [13]. Unfortunately, despite their structural diversity, these compounds are substrates of the AMEs to some extent and some of them even lost their antibiotic activity. More recently, we have demonstrated that the A site shows a clear tolerance for modification at the N-3″ position of the aminoglycosides [14,15] and that the guanidinylation of this position maintains the antibiotic activity against aminoglycoside 6′-acetyl-transferase and 4′-nucleotidyl-transfersase-expressing strains (Figure 1h) [16, 17].
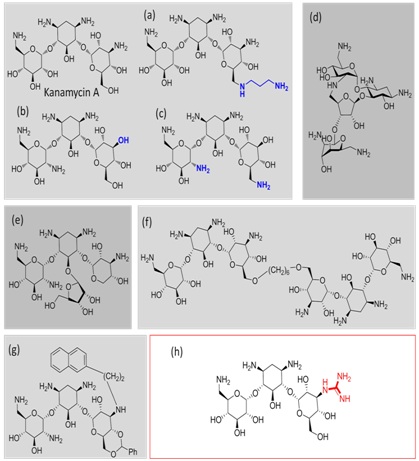
Figure 1: Representation of the different aminoglycoside systems (a-g) designed and synthesized by our group to prevent AME inactivation. Addition of a substituent at the N-3″ position of kanamycin A (strategy highlighted with a red square) is well tolerated by the rRNA and, particularly, guanidinylation of this position prevents the inactivation of the antibiotic by AMEs.
Herein, we propose the synthesis and evaluation of the new gentamicin derivatives 1a and 1b with the aim of expanding the usefulness of our strategy, demonstrating its broad applicability against other resistance enzymes. Gentamicin is a mixture of various congeners: C1, C1a and C2 (Figure 2). Structural differences between them are minor, differing only by a methyl or a hydrogen substitution in two R groups (R1 or R2) on the purpurosamine residue. This mixture has been the only aminoglycoside antibiotic used to date in clinic and recently it has demonstrated utility for the treatment of sepsis caused by diverse strains of MRSA bacteria [18]. The MIC of gentamicin (2a-c) typically ranges from 6 to 48 ?g/mL. However, many strains have acquired aminoglycoside resistance genes that encode various AMEs, which eventually result in very high resistance to gentamicin (MICs usually higher than 200 ?g/mL). The most clinically relevant of these genes are aac (6′), aac(2′), aph(2″) and ant(2″).
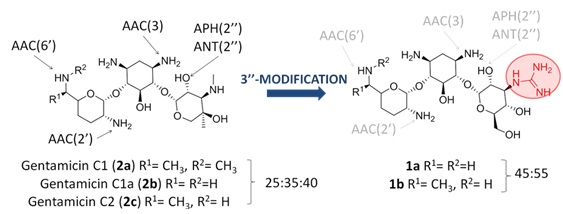
Figure 2: (Left) the structures of gentamicin C1, C1a and C2 components (2a-c) are shown, indicating the positions susceptible to enzymatic modification.
MATERIALS AND METHODS
Gentamicin (2a-c) was purchased from Sigma Aldrich. All the reactions that needed dry conditions were carried out under an argon atmosphere and the solvents were appropriately dried before use by standard techniques. Commercial reagents were used as received. All reactions were magnetically stirred and monitored by TLC in precoated Kiesel gel 60 F254 (Merck). Flash Column chromatography (FC) was carried out on Silica Gel 60 (32-63 μm). Detection was first carried out by UV light (254 nm) and then by charring with a solution of sulfuric acid, ninhydrin or with Mostain. NMR experiments were recorded on a Varian Unity 400 MHz at 313 K. The aminoglycosides 1a-1b were characterized employing a combination of 2D TOCSY, COSY and HSQC experiments. Low and high resolution mass spectra were provided by the Mass Spectrometry Facilities, CSIC in Madrid. The bacterial strains utilized in this study were obtained from the Hospital de Fuenlabrada (Madrid) and from ATCC (American Type Culture Collection) or CECT (Spanish Type Culture Collection).
SYNTHESIS OF COMPOUNDS 3A-3B
1,3,2′,6′,3″-(Cbz)5-gentamicin C1 (3a), 1,3,2′,6′,3″-(Cbz)5-gentamicin C1a (3b) and 1,3,2′,6′,3″ -(Cbz)5-gentamicin C2 (3c).
To a well stirred solution of gentamicin C1 (2a), C1a (2b) and C2 (2c) (ratio = 25:35:40) free base (0.3 g, 0.64 mmol) in a saturated aqueous solution of Na2CO3 (4 ml) at 0°C was added a solution of CbzCl (benzyl chloroformate) (0.65 g, 3.84 mmol) in acetone (1.5 ml) drop by drop. The mixture was vigorously stirred for 2 hours at this temperature and then 8 hours at room temperature. Subsequently, the solvent was removed under reduced pressure and the residue was pulverized in a mortar. This solid was added to an aqueous solution of HCl (1M) until neutralization and the formed solid product was filtered and dried under vacuum, obtaining the mixture of compounds 3a-c (0.36 g, 50%). MS-API-ES: 1148 (M+H)+ (3a), 1134 (M+H)+ (3b), 1120 (M+H)+ (3c).
1,3, 2′,6′,-(Cbz)4-2″,3″-carbamate-gentamicin C1 (4a), 1,3,2′,6′,-(Cbz)4-2″,3″-carbamate-gentamicin C1a (4b) and 1,3, 2′,6′,-(Cbz)4-2″,3″-carbamate-gentamicin C2 (4c).
To a stirred solution of the mixture of compounds 3a-c (0.3g, 0.32 mmol) in a mixture of 1,4-dioxane/H2O (30 ml/10 ml) was added an aqueous solution of NaOH (2.5M). The mixture was stirred at 50°C for 24 hours, after which was added an aqueous solution of HCl (1M) until pH 10. The mixture was concentrated under reduced pressure and subsequently water was added (25 ml). The obtained suspension was filtered and the solid was dried under vacuum. Finally, the residue was purified through a column chromatography (CHCl3/MeOH/NH4OH, 8:2:0.2 → 6:3:1), yielding the mixture of products 4a, 4b and 4c as a white solid (60%). MS-API-ES: 1040 (M+H)+ (4a), 1026 (M+H)+ (4b), 1012 (M+H)+ (4c).
1,3,2′,6′-(Cbz)4-gentamicin C1a (5a), 1,3,2′,6′-(Cbz)4-gentamicin C2 (5b) and 1,3,2′-(Cbz)3-gentamicin C1 (6).
To a solution of free base gentamicin C1, C1a and C2 (2a-c) (ratio = 25:35:40) (0.3 g, 0.64 mmol) in DMSO (4 ml) at 0°C was added N-benzyloxycarbonyl succinimide (0.52 g, 2.11 mmol). The mixture was stirred vigorously for 2 hours at room temperature. Subsequently, Et2O was added (100 ml), observing the formation of a colorless oil. After solvent decantation the residue was purified by resin chromatography column Amberlite™ IRA-120-H+ (5 g), yielding a mixture of compounds 5a and 5b (0.17 g, 37%) when the resin was eluted with a 0.5M solution of NH4OH in 1,4-dioxane/H2O (1:1) and compound 6 (73 mg, 13%) when the concentration of NH4OH was increased to 1M. MS-API-ES: 985 (M+H)+ (5a), 1000 (M+H)+ (5b) and MS-API-ES: 880 (M+H)+ (6).
1,3,2′,6′-(Cbz)4-3″-(Boc)2-guanidine-gentamicin C1a (7a) and 1,3,2′,6′-(Cbz)4-3″-(Boc)2-guanidinio-gentamicina C2 (7b).
To a stirred solution of compounds 5a and 5b (0.17 g, 0.23 mmol) in 1,4-dioxane (11.7 ml) was added 1,3-(Boc)2-2-(trifluoromethylsulfonyl)guanidine (0.134 g, 0.34 mmol) and Et3N (95 ? L, 0.69 mmol). The mixture was stirred at room temperature for 5 days. Then, the solvent was removed under reduced pressure and finally the residue was purified in silica gel column chromatography (AcOEt →AcOEt/MeOH 9:1) to yield a mixture of 7a and 7b (0.20 g, 72%) as a white foam. 1H NMR (DMSO-d6, 400 MHz) (selected signals): 7.60-7.20 (m, 40H), 5.35-5.27 (m, 4H), 5.25-4.95 (m, 10H), 2.10-1.70 (m, 4H), 1.50-1.40 (s, 18H), 1.20 (s, 6H). MS-API-ES: 1228 (M+H)+ (7a), 1244 (M+H)+ (7b).
3″-guanidine-gentamicin C1a (1a) and 3″-guanidine-gentamicin C2 (1b).
To a solution of compounds 7a and 7b (0.20 g, 0.165 mmol) in MeOH (3.2 ml) was added palladium on carbon (37 mg, 20% w/w) and acetic acid (1.0 ml). The reaction flask was purged three times and the mixture was stirred under a H2 atmosphere overnight, then filtered over a Celite® pad, washed with methanol and concentrated to dryness. The crude residue was used in the following reaction without further purification. Finally, this material was subjected to deprotection under acidic conditions by dissolving it in DCM/TFA (2.5 ml, 4:1 v/v). The reaction mixture was stirred at r.t. for 5 h, then evaporated to dryness and co-evaporated twice with toluene. The residue thus obtained was dissolved in distilled water, and the clear supernatant was taken and freeze-dried to yield a white fluffy powder 1a and 1b (67 mg, 82% two steps) as their corresponding TFA salts. 1H-NMR (D2O, 400 MHz): ? 5.19 (d, J = 3.6, 2H), 5.05 (d, J = 3.9 Hz, 2H), 3.83 – 3.79 (m, 2H), 3.79 – 3.61 (m, 6H), 3.47-3.24 (m, 6H), 3.16 (t, J = 5.0 Hz, 2H), 3.02 (t, J = 5.8 Hz, 2H), 2.82 – 2.72 (m, 5H), 2.78 (s, 3H), 2.68 (s, 3H), 2.52 – 2.21 (m, 2H), 1.68-1.48 (m, 4H), 1.48 - 1.39 (m, 2H), 1.19 (d, J = 6.8 Hz, 3H), 1.16 (d, J = 6.9 Hz). 13C-NMR (D2O, 100 MHz): ? 157.5, 103.1, 90.1, 89.5, 89.3, 77.1, 76.0, 75.1, 72.9, 72.0, 70.4, 67.0, 53.4, 52.5, 52.3, 52.2, 52.1, 51.9, 51.5, 47.5, 38.3, 39.5, 30.1, 29.2, 28.5, 27.7, 25.2, 24.2, 20.3. MS-API-ES (m/z): 492 (M+H)+ (1a), 506 (M+H)+ (1b). HRMS (m/z) 461.5631 (C19H39N7O6, 461.5640) (1a), 475.3115 (C20H41N7O6, 475.3118) (1b).
To a well stirred solution of gentamicin C1 (2a), C1a (2b) and C2 (2c) (ratio = 25:35:40) free base (0.3 g, 0.64 mmol) in a saturated aqueous solution of Na2CO3 (4 ml) at 0°C was added a solution of CbzCl (benzyl chloroformate) (0.65 g, 3.84 mmol) in acetone (1.5 ml) drop by drop. The mixture was vigorously stirred for 2 hours at this temperature and then 8 hours at room temperature. Subsequently, the solvent was removed under reduced pressure and the residue was pulverized in a mortar. This solid was added to an aqueous solution of HCl (1M) until neutralization and the formed solid product was filtered and dried under vacuum, obtaining the mixture of compounds 3a-c (0.36 g, 50%). MS-API-ES: 1148 (M+H)+ (3a), 1134 (M+H)+ (3b), 1120 (M+H)+ (3c).
1,3, 2′,6′,-(Cbz)4-2″,3″-carbamate-gentamicin C1 (4a), 1,3,2′,6′,-(Cbz)4-2″,3″-carbamate-gentamicin C1a (4b) and 1,3, 2′,6′,-(Cbz)4-2″,3″-carbamate-gentamicin C2 (4c).
To a stirred solution of the mixture of compounds 3a-c (0.3g, 0.32 mmol) in a mixture of 1,4-dioxane/H2O (30 ml/10 ml) was added an aqueous solution of NaOH (2.5M). The mixture was stirred at 50°C for 24 hours, after which was added an aqueous solution of HCl (1M) until pH 10. The mixture was concentrated under reduced pressure and subsequently water was added (25 ml). The obtained suspension was filtered and the solid was dried under vacuum. Finally, the residue was purified through a column chromatography (CHCl3/MeOH/NH4OH, 8:2:0.2 → 6:3:1), yielding the mixture of products 4a, 4b and 4c as a white solid (60%). MS-API-ES: 1040 (M+H)+ (4a), 1026 (M+H)+ (4b), 1012 (M+H)+ (4c).
1,3,2′,6′-(Cbz)4-gentamicin C1a (5a), 1,3,2′,6′-(Cbz)4-gentamicin C2 (5b) and 1,3,2′-(Cbz)3-gentamicin C1 (6).
To a solution of free base gentamicin C1, C1a and C2 (2a-c) (ratio = 25:35:40) (0.3 g, 0.64 mmol) in DMSO (4 ml) at 0°C was added N-benzyloxycarbonyl succinimide (0.52 g, 2.11 mmol). The mixture was stirred vigorously for 2 hours at room temperature. Subsequently, Et2O was added (100 ml), observing the formation of a colorless oil. After solvent decantation the residue was purified by resin chromatography column Amberlite™ IRA-120-H+ (5 g), yielding a mixture of compounds 5a and 5b (0.17 g, 37%) when the resin was eluted with a 0.5M solution of NH4OH in 1,4-dioxane/H2O (1:1) and compound 6 (73 mg, 13%) when the concentration of NH4OH was increased to 1M. MS-API-ES: 985 (M+H)+ (5a), 1000 (M+H)+ (5b) and MS-API-ES: 880 (M+H)+ (6).
1,3,2′,6′-(Cbz)4-3″-(Boc)2-guanidine-gentamicin C1a (7a) and 1,3,2′,6′-(Cbz)4-3″-(Boc)2-guanidinio-gentamicina C2 (7b).
To a stirred solution of compounds 5a and 5b (0.17 g, 0.23 mmol) in 1,4-dioxane (11.7 ml) was added 1,3-(Boc)2-2-(trifluoromethylsulfonyl)guanidine (0.134 g, 0.34 mmol) and Et3N (95 ? L, 0.69 mmol). The mixture was stirred at room temperature for 5 days. Then, the solvent was removed under reduced pressure and finally the residue was purified in silica gel column chromatography (AcOEt →AcOEt/MeOH 9:1) to yield a mixture of 7a and 7b (0.20 g, 72%) as a white foam. 1H NMR (DMSO-d6, 400 MHz) (selected signals): 7.60-7.20 (m, 40H), 5.35-5.27 (m, 4H), 5.25-4.95 (m, 10H), 2.10-1.70 (m, 4H), 1.50-1.40 (s, 18H), 1.20 (s, 6H). MS-API-ES: 1228 (M+H)+ (7a), 1244 (M+H)+ (7b).
3″-guanidine-gentamicin C1a (1a) and 3″-guanidine-gentamicin C2 (1b).
To a solution of compounds 7a and 7b (0.20 g, 0.165 mmol) in MeOH (3.2 ml) was added palladium on carbon (37 mg, 20% w/w) and acetic acid (1.0 ml). The reaction flask was purged three times and the mixture was stirred under a H2 atmosphere overnight, then filtered over a Celite® pad, washed with methanol and concentrated to dryness. The crude residue was used in the following reaction without further purification. Finally, this material was subjected to deprotection under acidic conditions by dissolving it in DCM/TFA (2.5 ml, 4:1 v/v). The reaction mixture was stirred at r.t. for 5 h, then evaporated to dryness and co-evaporated twice with toluene. The residue thus obtained was dissolved in distilled water, and the clear supernatant was taken and freeze-dried to yield a white fluffy powder 1a and 1b (67 mg, 82% two steps) as their corresponding TFA salts. 1H-NMR (D2O, 400 MHz): ? 5.19 (d, J = 3.6, 2H), 5.05 (d, J = 3.9 Hz, 2H), 3.83 – 3.79 (m, 2H), 3.79 – 3.61 (m, 6H), 3.47-3.24 (m, 6H), 3.16 (t, J = 5.0 Hz, 2H), 3.02 (t, J = 5.8 Hz, 2H), 2.82 – 2.72 (m, 5H), 2.78 (s, 3H), 2.68 (s, 3H), 2.52 – 2.21 (m, 2H), 1.68-1.48 (m, 4H), 1.48 - 1.39 (m, 2H), 1.19 (d, J = 6.8 Hz, 3H), 1.16 (d, J = 6.9 Hz). 13C-NMR (D2O, 100 MHz): ? 157.5, 103.1, 90.1, 89.5, 89.3, 77.1, 76.0, 75.1, 72.9, 72.0, 70.4, 67.0, 53.4, 52.5, 52.3, 52.2, 52.1, 51.9, 51.5, 47.5, 38.3, 39.5, 30.1, 29.2, 28.5, 27.7, 25.2, 24.2, 20.3. MS-API-ES (m/z): 492 (M+H)+ (1a), 506 (M+H)+ (1b). HRMS (m/z) 461.5631 (C19H39N7O6, 461.5640) (1a), 475.3115 (C20H41N7O6, 475.3118) (1b).
MIC DETERMINATION
A solution of the selected bacterial strain was grown in 1 ml of Mueller-Hinton broth to an optical density (OD600) of 0.5units. The desired concentrations of guanidino-glycosides 1a-b were added from stock solutions. After incubation at 37°C for 24 h, the OD600 of each sample was read in duplicate. In both cases, the MIC was taken as the lowest antibiotic concentration inhibiting bacterial growth by greater than 90%.
In the case of the resistance strain expressing AAC (6′)-Ib, E. coli BL21 (DE3) containing the pET-AAC(6′)-Ib plasmid, enzyme production was induced with IPTG prior to addition of the antibiotics.
In the case of the resistance strain expressing AAC (6′)-Ib, E. coli BL21 (DE3) containing the pET-AAC(6′)-Ib plasmid, enzyme production was induced with IPTG prior to addition of the antibiotics.
RESULTS AND DISCUSSION
Chemistry
Recently, we have described the synthesis of 3″-guanidino kanamycin A by regioselective 3″-amino deprotection of tetra-Cbz-protected kanamycin A under basic conditions and subsequent guanidinylation [16]. Using a similar approach (Figure 3) 1,3,6′,2′-3″-(Cbz)5-gentamicins (3a-c) were obtained by treatment of gentamicin (2a-c) with benzyl chloroformate in a Na2CO3 saturated aqueous solution and acetone mixture. However, when 3a-c were reacted with a 2.5M solution in a 1,4-dioxane and H2O mixture at 50°C [19], the carbamate of position 3″ cyclized, obtaining a mixture of urethanes 4a-c as expected, but unfortunately these intermediates were too stable and did not undergo ring opening. The use of higher temperature and/or longer times and/or a higher concentration of base and/or a stronger base such as KOH at different concentrations caused side reactions with concomitant chemical decomposition of the starting material.
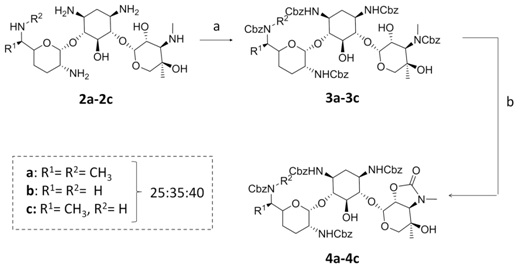
Figure 3: Reagents and conditions: (a) CbzCl, Acetone/Na2CO3 aq. sat., 0°C, 10 h, 50%; (b) NaOH (2.5M), 1,4-dioxane/H20, 50°C, 24 h, 60%.
In view of these results a slightly different approach was employed (Figure 4). Treatment of gentamicin 2a-c with N-(benzyloxycarbonyloxy)succinimide in DMSO afforded a mixture of 6′,2′,1,3-(Cbz)4-gentamicin C1a (5a) and 6′,2′,1,3-(Cbz)4-gentamicin C2 (5b) in 37% (45:55) which was employed in the next reaction and 2′,1,3-(Cbz)3-gentamicin C1 (6) in 13% yield that was discarded [20]. Conversion of 3″-methyl amino groups to the corresponding guanidine was achieved by treatment of the mixture (5a-b) with 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine and Et3N [16], obtaining 6′,2′,1,3-(Cbz)4-3″-guanidino(Boc)2 gentamicins C1 and C2 (7a-b) in 72% yield. Catalytic hydrogenolysis of the Cbz groups and finally, acidic deprotection of the guanidine Boc groups, afforded a mixture of 3″-guanidino-gentamicins C1 and C2 (1a-b) in 82% yield as their TFA salt (Figure 4).
Figure 3: Reagents and conditions: (a) CbzCl, Acetone/Na2CO3 aq. sat., 0°C, 10 h, 50%; (b) NaOH (2.5M), 1,4-dioxane/H20, 50°C, 24 h, 60%.
In view of these results a slightly different approach was employed (Figure 4). Treatment of gentamicin 2a-c with N-(benzyloxycarbonyloxy)succinimide in DMSO afforded a mixture of 6′,2′,1,3-(Cbz)4-gentamicin C1a (5a) and 6′,2′,1,3-(Cbz)4-gentamicin C2 (5b) in 37% (45:55) which was employed in the next reaction and 2′,1,3-(Cbz)3-gentamicin C1 (6) in 13% yield that was discarded [20]. Conversion of 3″-methyl amino groups to the corresponding guanidine was achieved by treatment of the mixture (5a-b) with 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine and Et3N [16], obtaining 6′,2′,1,3-(Cbz)4-3″-guanidino(Boc)2 gentamicins C1 and C2 (7a-b) in 72% yield. Catalytic hydrogenolysis of the Cbz groups and finally, acidic deprotection of the guanidine Boc groups, afforded a mixture of 3″-guanidino-gentamicins C1 and C2 (1a-b) in 82% yield as their TFA salt (Figure 4).
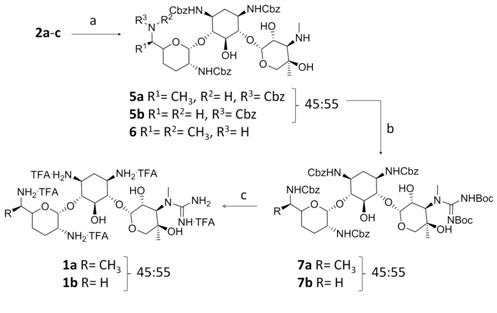
Figure 4: Reagents and conditions: (a) N-(benzyloxycarbonyloxy)succinimide, DMSO, 0°C, 2h, 37% (5a and 5b) and 13% (6); (b) 1,3-di-Boc-2-(trifluoromethylsulfonyl)guanidine, Et3N, 1,4-dioxane, 5 d, 72%; (c) i. H2, Pd•C, AcOH, MeOH, 12 h. ii. TFA/DCM (1:4), r.t., 5 h, 82% (two steps).
Antibacterial activities
In order to determine the biological activity of 3″-guanidino-gentamicin (1a-b), Minimum Inhibitory Concentration Experiments (MICs) were carried out against a wide variety of bacterial strains (Figure 5). According to our results, the mixture of gentamicin congeners (2a-c) is a powerful antibiotic against E. coli (strain ATCC 25922) (MIC= 0.5 ?g/ml), which is in accordance with results previously reported [21]. However, when AAC (6′)-Ib is expressed, alone or in combination with APH (2″) or APH(3′), gentamicins (2a-c) completely lose the antibiotic activity (MIC > 250 ?g/ml). To our delight, 3″-guanidino-gentamicins (1a-b) still maintained a significant antibiotic activity against E.coli and more importantly, against resistant strains expressing AAC (6′)-Ib, APH (2″) and APH (3′). Finally, we tested the effect of 1a-b against ANT (6), an aminoglycoside resistance enzyme which is only efficient on streptomycin. We chose this particular enzyme since previous (unpublished) results from our group showed that the introduction of guanidine moieties on kanamycin A turns this scaffold into a ligand (not a substrate) of this enzyme. According to these results, we decided to test the new antibiotic against this enzyme to demonstrate that this modification does not transform gentamicin into a substrate either.
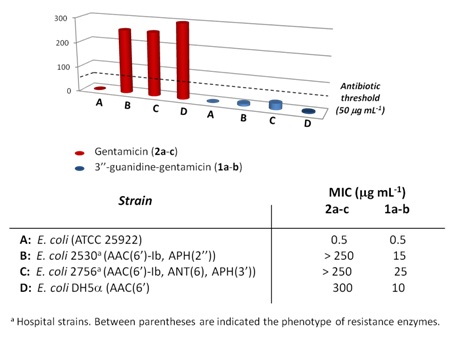
Figure 5: MIC values of commercial gentamicin (2a-c) (red) and novel 3″-guanidine gentamicin (1a-b) (blue). The antibiotic threshold (50??g/ml) is shown with a dashed line.
CONCLUSION
In the present study, we have synthesized two new 3″-guanidino gentamicin derivatives. The guanidinylation at the N-3″ position not only maintains the biological activity against a simple E.coli strain, but also against a representative set of resistant strains expressing several AMEs (AAC(6′)-Ib, APH(2″), ANT(6′) and APH(3′)). This result provides an excellent starting point for the development of semisynthetic next generation compounds effective in the presence of AMEs and it is a confirmation of our previous results on regioselective modification of position 3″ of aminoglycoside antibiotics.
ACKNOWLEDGEMENT
This work was supported by the Spanish Ministerio de Economía y Competitividad (Grant MAT2015-65184-C2-2-R) and CSIC project iCOOPB20237.
REFERENCES
- Waskman SA, Bugie E, Schatz A (1944) Isolation of antibiotic substances from soil microorganisms, with special reference to streptothricin and streptomycin. Proc Staff Meet Mayo Clin 19: 537-548.
- Kling D, Chow C, Mobashery S (2007) Binding of Antibiotics to the Aminoacyl tRNA Site of Bacterial Ribosome. In: Dev P Arya (ed.). Aminoglycoside Antibiotics: from Chemical Biology to Drug Discovery, Wiley, New York, USA.
- Vicens Q, Westhof E (2003) RNA as a drug target: The Case of Aminoglycosides. ChemBioChem 4: 1018-1023.
- Walter F, Vicens Q, Westhof E (1999) Aminoglycoside-RNA interactions. Curr Opin Chem Biol 3: 694-704.
- target="_blank"Mingeot-Leclercq MP, Tulkens PM (1999) Aminoglycosides: Nephrotoxicity. Antimicrob Agents Chemother 43: 1003-1012.
- Garneau-Tsodikova S, Labby KJ (2016) Mechanisms of Resistance to Aminoglycoside Antibiotics: Overview and Perspectives. MedChemComm 7: 11-27.
- Chandrika NT, Garneau-Tsodikova S (2016) A review of Patents (2011-2015) towards combating Resistance and Toxicity of Aminoglycosides. Med Chem Commun 7: 50-68.
- Santana AG, Zarate, SG, Bastida A, Revuelta J (2015) Targeting RNA with Aminoglycosides: Current Improvements in their Synthesis and Biological Activity. In. Atta-ur Rahman, M. Iqbal Choudhary MI (eds.). Frontiers in Anti-infective drug discovery. Bentham Science, Emirate of Sharjah, United Arab Emirates.
- Revuelta J, Vacas T, Torrado M, Corzana F, Gonzalez C, et al. (2008) NMR-Based analysis of aminoglycoside recognition by the resistance enzyme ANT(4′): The pattern of OH/NH3+ substitution determines the preferred antibiotic binding mode and is critical for drug inactivation. J Am Chem Soc 130: 5086 -5103.
- Bastida A, Hidalgo A, Chiara JL, Torrado M, Corzana F, et al. (2006) Exploring the Use of Conformationally Locked Aminoglycosides as a New Strategy to Overcome Bacterial Resistance. J Am Chem Soc 128: 100-116.
- Revuelta J, Vacas T, Corzana F, Gonzalez C, Bastida A, et al. (2010) Structure-based design of highly crowded ribostamycin/kanamycin hybrids as a new family of antibiotics. Chem Eur J 16: 2986-2991.
- Santana AG, Bastida A, Martínez del Campo T, Asensio JL, et al. (2011) An efficient and general route to the synthesis of novel aminoglycosides for RNA binding. Synlett 2: 219-222.
- Vacas T, Corzana F, Jiménez-Osés G, González C, Gómez AM, Bastida A, et al. (2010) Role of aromatic rings in the molecular recognition of aminoglycoside antibiotics: Implications for drug design. J Am Chem Soc 132: 12074-12090.
- Jiménez-Moreno E, Gómez-Pinto I, Corzana F, Santana AG, Revuelta J, et al. (2013) Chemical interrogation of drug/RNA complexes: From chemical reactivity to drug design. Angew Chem Int Ed 52: 3148-3151.
- Jiménez-Moreno E, Montalvillo-Jiménez L, Santana AG, Gómez AM, Jiménez-Osés G, et al. (2016) Finding the right candidate for the right position: A fast NMR-assisted combinatorial method for optimizing nucleic acids binders. J Am Chem Soc 138: 6463-6474.
- Santana AG, Zárate SG, Asensio JL, Revuelta J, Bastida A (2016) Selective modification of the 3″-amino group of kanamycin prevents significant loss of activity in resistant bacterial strains. Org Biomol Chem 14: 516-525.
- Streicher W, Loibner H, Hildebrandt J, Tunrowsky F (1983) Synthesis and Structure/Activity Relationships of New Guanidino Derivatives of Aminoglycoside Antibiotics. Drugs Exp Clin Res 9: 591-598.
- Gonzalez-Padilla M, Torre-Cisneros J, Rivera-Espinar F, Pontes-Moreno A, López-Cerero L, et al. (2015) Gentamicin therapy for sepsis due to carbapenem-resistant and colistin-resistant Klebsiella pneumonia. J Antimicrob Chemother 70: 905-913.
- Chen G-H, Pan P, Chen Y, Meng X-B, Li Z-J (2009) Selective deprotection of the Cbz amine protecting group for the facile synthesis of kanamycin A dimers linked at N-3'' Tetrahedron 65: 5922-5927.
- Moon MS, Jun SJ, Lee SH, Cheong CS, Kim KS, et al. (2005) A semi synthesis of isepamicin by fragmentation method. Tetrahedron Lett 46: 607-609.
- Jakobsen L, Sandvang D, Jensen VF, Seyfarth AM, Frimodt-Møller N, et al. (2007) Gentamicin susceptibility in Escherichia coli related to the genetic background: problems with breakpoints. Clin Microbiol Infect 13: 830-832.
Citation: Zárate SG, Bastida A, Santana AG, Revuelta J (2017) Recovery of the Antibiotic Activity against Resistant Bacterial Strains by Selective Guanidinylation of the 3″-Methylamino Group of Gentamicin. J Mod Chem Sci 1: 002.
Copyright: © 2017 Julia Revuelta, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!