Renal involvement in IgG4-related disease (IgG4-RD). Why is it so Important?
*Corresponding Author(s):
María Carmen Prados SolerNephrology Department, Torrecárdenas, University Hospital, Almería, Spain
Email:lensasu@yahoo.es
Abstract
Immunoglobulin G4-Related Disease (IgG4-RD) is a recently recognized recurrent multisystem fibro inflammatory pseudo-tumor-forming entity that includes multiple diseases previously considered idiopathic and organ-specific, including several forms of renal involvement. We present a case of a 71-year-old man with a clinical picture that began in 2016, when he presented with autoimmune pancreatitis. Initially, it was thought that the patient had pancreatic cancer, so it was necessary to perform a pancreatic biopsy. The patient was referred to our clinic 4 years later due to creatinine 2.2 mg/dl, elevated IgG4 and C3-C4 hypocomplementemia, so a renal biopsy was scheduled to confirm IgG4-RD. This showed, on light microscopy with hematoxylin-eosin, increased cellularity in the interstice with a dense chronic inflammatory infiltrate, at the expense of plasma cells, expanding and fibrotic. Immunohistochemistry showed 14 plasma cells per high magnification field expressing IgG4, confirming the clinical diagnosis of severe interstitial involvement in the context of IgG4-RD. The patient was treated with corticosteroids, presenting several relapses. He was not treated with rituximab because he was already considered to have poor renal functional reserve.
Keywords
Immunoglobulin G4-related disease (IgG4-RD); Relapses; Rituximab
Introduction
IgG4-RD is a recently recognized recurrent multisystem fibroinflammatory pseudo-tumor-forming entity that includes multiple diseases previously considered idiopathic and organ-specific, including several forms of renal involvement [1]. The common features shared by the different entities that make up IgG4-RD are: elevated serum IgG4 levels, alterations in imaging tests with neoplastic-like swelling of the affected organs, histopathological and immunostaining features, and good response to treatment with glucocorticoids [2].
IgG4-RD has now been described in virtually every organ besides the pancreas, including the bile duct, salivary glands, periorbital tissues, pituitary gland, kidneys, lungs, lymph nodes, meninges, breast, prostate, thyroid, pericardium and skin, with similar histological features [3].
Autoimmune Pancreatitis (AIP) is a benign fibroinflammatory disease of the pancreas first described in 1961 as a case of pancreatitis associated with hypergammaglobulinemia. In 1995, Yoshida et al. proposed the concept of AIP. The disease was associated with the presence of serum IgG4 in 2001. Subsequently, in 2003, patients with type 1 AP were found to have IgG4-rich fibroinflammatory lesions, both synchronous and metachronous, in other organs. This expanded the concept from local disease to multi-organ or systemic disease [4]. The first references to the kidney date back to 2004 in Japan, where a case of non-granulomatous Tubulointerstitial Nephritis (TIN) in a patient with IgG4-related AIP was published. At the Boston International Symposium in October 2011, the term IgG4-RD was proposed and in 2012 the first consensus document on the disease was produced [5].The diagnosis of these diseases requires a high index of suspicion and renal biopsy may be the key to the diagnosis of this entity. Response to corticosteroids is the rule [6]. Biological therapy with rituximab has been used successfully to induce and sustain clinical response and is therefore proposed as a second-line therapy. It is important to be aware of this disease, to suspect it and to make an early and accurate diagnosis.
Clinical Case
71-year-old male, ex-smoker with a history of hypertension, diabetes mellitus type 2, irritable bowel syndrome and benign prostatic hyperplasia. In 2016 he was admitted to other Hospital in Granada due to obstructive jaundice, with initial suspicion of pancreatic cancer. After pancreatic biopsy, he was diagnosed with AIP associated with IgG4 (Type 1) and a 5mm pancreatic cyst at the corpus-tail junction, requiring a biliary stent for a 1 month. Spectacular response to corticoids. Referred to the Nephrology Department in April/2020 due to progressive deterioration of renal function (Cr 1.2 → 2.8 → 3.7 mgr/dl), negative proteinuria and absence of hematuria. Laboratory tests showed hypocomplementemia (C3: 40 and C4<7) and IgG 2584. Other immunological tests were negative. At that time he was taking prednisone 2.5 mgr/day.
With the suspicion of IgG4-RD, an ultrasound-guided renal biopsy was scheduled. The sample contained 18 glomeruli, 16 of which were globally sclerosed. The non-sclerosed glomeruli showed fibrosis of Bowman´s capsule and an ischemic appearance. In the interstice, an intense inflammatory infiltrate consisting of T lymphocytes (CD3), to a lesser extent B (CD 20+), numerous CD 68+ macrophages and a percentage of plasma cells, with expression of both kappa light chain and lambda, most of them IgG, was very striking. IgG4 staining showed 10-14 positive cells in the high magnification fields with the highest number of cells. This inflammatory infiltrate was associated with significant diffuse fibrosis of a somewhat storiform appearance producing a striking loss of tubular structures, with few scattered atrophic tubules remaining. The arteries showed moderate to severe intimal thickening. Cytomegalovirus and SV40 stains were negative. Ebstein-Barr negative. Congo red stain negative. The immunofluorescence sample contained 7 glomeruli of which 4 were sclerosed, granular basement membrane deposits were observed for IgG, kappa chain, lambda, C3, C1q, C4d and C4. The definitive diagnosis was tubulointerstitial nephritis with moderate focal increase of IgG4 plasma cells and extensive fibrosis with a storiform appearance. Severe glomerular sclerosis. (Figures 1-5)
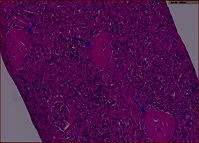 Figure 1: HE x20. Glomerular sclerosis.
Figure 1: HE x20. Glomerular sclerosis.
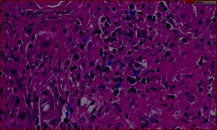 Figure 2: HE x60. Intesticial inflammatory infiltrate with Plasma cell component.
Figure 2: HE x60. Intesticial inflammatory infiltrate with Plasma cell component.
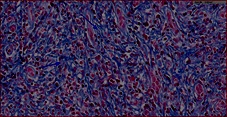 Figure 3: Masson's trichome x 40. Fibrosis.
Figure 3: Masson's trichome x 40. Fibrosis.
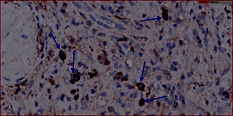 Figure 4: IgG4 x 60. Positivity for IgG4 elements.
Figure 4: IgG4 x 60. Positivity for IgG4 elements.
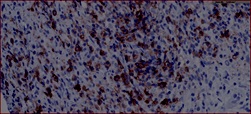 Figure 5: CD 38 x 40. Stain revealing the plasma cell component.
Figure 5: CD 38 x 40. Stain revealing the plasma cell component.
The patient received treatment with MP iv 3 boluses of 500 mgr and then oral prednisone (60 mgr/day and then tapering), with improvement of renal function (Cr 2.2 mgr/dl) despite renal biopsy findings of poor prognosis. In May/2022, coinciding with dose reduction (5 mgr/day → 2.5 mgr/day), renal function worsens (Cr 2.2 mgrdl → 4.78 mgr/dl), starting again with prednisone 60 mgr/day and improving to Cr 3 mgr/dl – Chronic Kidney Disease stage 4. In October/2022, without any triggering factor, he attended the Emergency Department, presenting with new deterioration of renal function (Cr 3.5 → 6 mgr/dl). On this date, IgG was normal. Faced with this situation, two attitudes could be taken: consider the patient in a situation of advanced chronic kidney disease, in a pre-dialysis situation, or the opposite, try again to improve renal function. It was decided to administer 3 boluses of 500 mgr iv MP again and to continue with prednisone 40 mgr/day in a descending pattern. He was not treated with rituximab because he was considered to have advanced chronic kidney disease. Against all prognosis, we achieved an improvement in renal function to Cr 3 mgr/dl. He is currently hospitalized for polyarthritis and Staphylococcus aureus abscess in the left lower limb, requiring drainage and cloxacilin iv. Renal function stable (Cr 3 mgr/dl), 2 months after the last outbreak with renal involvement.
Discussion
ER-IgG4 is a recently recognized immune-mediated, fibroinflammatory, multisystemic, recurrent, pseudo-tumor-forming, that affect almost any organ and often presents with multi-organ involvement, although the most commonly affected are the pancreas, salivary glands, lacrimal glands, lymph nodes, retroperitoneum and kidneys [7]. Three histopathological findings characterize the disease in the affected organs: 1) the presence of sclerosis in a storiform pattern; 2) a dense lymphoplasmacytic infiltrate; and 3) an increased ratio of IgG4-positive cells by immunohistochemistry to IgG-positive cells. The average of this ratio is a ratio of IgG4+/IgG+ cells greater than 40%, but the criterion varies according to the organ involved [8].
The mean age at diagnosis is approximately 60 years and there is a male predominance with a male: female ratio of 8:3, characteristics shared by our patient [9]. It includes multiple diseases that were previously considered idiopathic and organ-specific, such as autoimmune pancreatitis, Mikulicz disease, Riedel´s thyroiditis and Ormond´s disease, as well as various forms of renal involvement, such as membranous glomerulonephritis, Schönlein-Henoch purpura, IgA nephropathy, membranoproliferative glomerulonephritis, minimal change glomerulonephritis, amyloidosis amyloid A, obstructive renal failure in the context of urinary tract obstruction when retroperitoneal fibrosis is present, etc. However, the most common parenchymal lesion is tubulointerstitial nephritis, so in our case we must rule out pharmacological, infectious, idiopathic, neoplastic, metabolic disorders and those associated with systemic diseases, as it can be confused with Sjögren syndrome, although this has a clearly defined serological profile of autoimmunity or ANCA-associated vasculitis [9, 10]. In terms of laboratory findings, elevated serum levels of total IgG, IgG4 > 135 mg/dl and IgE >360 mgr/dl are common and have been reported. However, it is now recognized that serum IgG4 levels are normal in a substantial percentage of patients with a clinicopathological diagnosis of IgG4-RD. The sensitivity of serum IgG4 ranges from 50-90%, with a highly variable specificity as low as 60%. The existence of an elevated serum IgG4 level is no longer considered essential for the diagnosis of ER-IgG4. In fact, some organ systems and anatomical regions (e.g. the retroperitoneum) are less likely to be associated with elevated serum IgG4 that others. Other findings included hypocomplementemia, which has been identified in 25% of patients with ER-IgG4, especially in those with renal, pulmonary, aortic and submandibular gland involvement, and has been associated with increased activity [11-13].
Clinically, the differential diagnosis must be made with other systemic diseases such as systemic lupus erythematosus, Sjögren syndrome, cryoglobulinemia, granulomatosis with polyangiitis, sarcoidosis, diabetic nephropathy, drug-induced TIN, idiopathic membranous glomerulonephritis and necrotizing glomerulonephritis pauci-immune [14]. Radiologically, the differential diagnosis is also broad and depends on the morphology of the lesions in the imaging tests. Thus, when renal involvement is nodular and bilateral, the differential diagnosis must be made with metastatic lesions, lymphoma, bilateral pyelonephritis and septic embolism, among others. If it is a single unilateral mass-like lesion it may simulate a renal cell carcinoma and if it manifests as a local thickening in the pelvis it may simulate a transitional cell carcinoma. If the lesion has a wedge morphology it may resemble a single focus of pyelonephritis or even a renal infarction [14]. The join analysis of clinical, serological and radiological features is necessary to narrow down the differential diagnosis, and to determine whether histological examination is necessary and from which organ to obtain it [15, 16]. In terms of treatment, it must be analyzed whether the patient requires is or not, with an affirmative response in the vast majority of cases. Arguments in favor include the better response and prognosis of the affected organ with early initiation of therapy, the possibility of multi-organ involvement that could benefit, and the generally favorable response that exists with therapy. Exceptions would include patients with minimally symptomatic disease or without major prognostic implications (isolated and non-limiting enlargement of salivary or lacrimal glands, adenopathies), in which other organ involvement is excluded, as well as patients with predominantly fibrous involvement, who benefit more from surgical than pharmacological intervention [17]. The disease usually responds to high doses of glucocorticoids, with an average of 40 mg/day and a gradual tapering off. It is, in fact, the response to corticosteroid therapy that is a further diagnostic criterion [18, 19]. However, the relapse rate during and after glucocorticoid tapering is high, so international consensus recommends a much more gradual tapering of the dose, over a period of 3-6 months. In our case, 6-methyl-prednislone was started at 1 mg/Kg/day and a dramatic clinical improvement was achieved, although he relapsed 3 times during his follow-up. Very favorable results were recently published with the use of rituximab as induction and/or maintenance therapy. In fact, many cases series suggest that B-cell depletion with rituximab could be an effective therapy to treat IgG4-RD especially for patients with multi-organ disease [20-22]. An open-label pilot trial with patients at highs risk of relapse, refractor disease due to multi-organ involvement and failure of prior therapy with conventional Disease-Modifying Antirheumatic Drugs (DMARDs), used rituximab as induction therapy at a dose of 1000 mg on days 1 and 15, without concurrent corticosteroids (26 patients) or with gradual tapering of corticosteroids over the first 2 months (4 patients), and 97% of individuals achieved response at 6 months, with 47% in complete remission [23,24].
Conclusion
To establish the diagnosis of IgG4-RD, the clinical imperative is to correlate clinical, serological, radiological and pathological data. None of these approaches alone provides definitive evidence for accurate patient classification. The diagnosis of this disease requires a high of suspicion and renal biopsy is undoubtedly fundamental, with immunohistochemistry techniques with IgG4 being key. Relapses are frequent when the steroid dose is reduced or treatment is stopped. Corticosteroids increase the risk of infections, as in the case of our patient with a lower limb abscess. High doses need to be administered and a slow tapering schedule for 6 months, even in some patients they can never be discontinued. The persistence of elevated IgG4 and/or the reappearance of hypocomplementemia should alert us to the possibility of relapse. B-cell depletion with rituximab may be an effective therapy. In our case, we did not consider treatment with rituximab to be indicated due to the stage of chronic kidney disease; perhaps it should have been administered much earlier. We considered adding mycophenolate mofetil as an anti-fibrotic.
References
- Brito-Zerón P, Ramos-Casals M, Bosch X, Stone J (2014) The clinical spectrum of IgG4-related disease. Autoimmun Rev 13:1203-1210.
- Matinez-Valle F, Orozco Galvez O, Fernandez-codina A (2018) Etiopathogenesis, diagnosis and treatment of IgG4-related disease. Med Clin (Barc) 151: 18-25.
- Wallace Z, Zhang, Perugino C, Naden R, Choi H, et al. (2019) Clinical phenotypes of IgG4-related disease: analysis of two international cross-sectional cohorts. Ann Rheum Dis 78: 406-412.
- Evans R, Cargill T, Goodchild G, Oliveira B, Rodriguez-Justo M, et al. (2019) Clinical manifestations and long-term outcomes of IgG4-related kidney and retroperitoneal involvement in a United Kingdom IgG4-related disease cohort. Kidney Int Rep 4: 48-58.
- Pradhan D, Pattnaik N, Silowash R, Mohanty SK (2015) IgG4-related kidney disease. A review. Pathol Res Pract 211: 707-711.
- Masaki Y (2014) Glucocorticoids for the treatment of IgG4-related disease. 2nd international IgG4-related disease symposium. Wakiki, Hawaii.
- Ryu JH, Horie R, Sekiguchi H, Peikert T, Yi ES (2012) Spectrum of disorders associated with elevated serum IgG4 levels encountered in clinical practice. In J Rheumatol 2012: 232960.
- Deshpande V, Chan JK, Yi EE, Sato Y, Yoshino T, et al. (2012) Consensus statement on the pathology of IgG4-related disease. Mod Pathol 25:1181-1192.
- Pieringer H, Parzer I, Wöhrer A, Reis P, Oppl B, et al. (2014) IgG4-related disease. An orphan disease with many faces. Orphanet J Rare Dis 9: 110.
- Kamisawa T, Zen Y, Pillai S, Stone JH (2015) IgG4-related disease. Lancet 385: 1460-1471.
- Carruthers NM, Khosroshahi A, Augustin T, Deshpande V, Stone JH (2015) The diagnostic utility of serum Ig G4 concentrations in IgG4-related disease. Ann Rheum Dis 74:14-8.
- Yamamoto M, Tabeya T, Naishiro Y, Yajima H, Ishigami K, et al. (2012) Value of serum IgG4 in the diagnosis of IgG4-related disease and in differentiation from rheumatic diseases and other diseases. Mod Rheumatol 22: 419-425.
- Wallace ZS, Deshpande V, Mattoo H, Mahajan VS, Kulikova M, et al. (2015) IgG4-related disease: Clinical and laboratory features in one hundred and twenty-five patients. Arthritis Rheumatol 67: 2466-2475.
- Legatowicz-Koprowska M (2018) IgG4-related disease: Why is it so important?. Cent Eur Immunol 43: 204-208.
- Wallace ZS, Naden RP, Chari S, Choi HK, Della-Torre E, et al. (2020) The 2019 American College of Rheumatology/European League Against Rheumatism classification criteria for IgG4-related disease. Ann Rheum Dis 79: 77-87.
- Ardila-Suarez O, Abril A, Gomez-Puerta J (2017) IgG4-related disease: a concise review of the literatura. Rheumatol Clin 13: 160-166.
- Carruthers MN, Stone JH, Deshpande V, Kosroshahi A (2012) Development of an IgG4-RD Responder Index. Int J Rheumatol 2012259408.
- Kamisawa T, Shimosegawa T, Okazaki K, Nishino T, Watanabe H, et al. (2009) Standard steroid treatment for autoimmune pancreatitis. Gut 58: 1504-1507.
- Sandanayake NS, Church NI, Chapman MH, Johnson GJ, Dhar DK, et al. (2009) Presentation and management of post-treatment relapse in autoimmune pancreatitis/inmmunoglobulin G4-associated cholangitis. Clin Gastroenterol Hepatol 7: 1089-1996.
- Krosrashahi A, Bloch DB, Deshpande V, Stone JH (2010) RTX therapy leads to rapid decline of serum IgG4 levels and prompt clinical improvement in IgG4-related systemic disease. Arthritis Rheum 62: 1755-62.
- Hart PA, Toazian MD, Witzig TE, Clain JE, Gleeson FC, et al. (2013) Treatment of relapsing autoimmune pancreatitis with immunomodulators and RTX: the Mayo Clinic experience. Gut 62:1607-1615.
- Carruthers M, Topazian M, Khosroshahi A, Witzig T, Wallace Z, et al. (2015) Rituximab for IgG4-related disease: a prospective, open-label trial. Ann Rheum Dis 74: 1171-1117.
- Yamamoto M, Awakawa T, Takahashi H (2015) Is rituximab effective for IgG4-related disease in the long term? Experience of cases treated with rituximab for 4 years. Ann Rheum Dis 74: e46.
- Khosroshahi A, Wallace Z, Crowe JL, Akamizu T, Azumi A, et al. (2015) International consensus guidance statement on the management and treatment of IgG4-related disease. Arthritis Rheumatol 67: 1688-1699.
Citation: María Carmen Prados Soler, María Dolores Salmerón Rodríguez, Isabel María Villegas Pérez, Marta Panadero Moya and Francisco Javier González Martínez (2023) Renal involvement in IgG4-related disease (IgG4-RD). Why is it so important?. J Nephrol Renal Ther 9: 074.
Copyright: © 2023 María Carmen Prados Soler, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.