Role of P38 MAPK in Bile Formation and Cholestasis
*Corresponding Author(s):
Anwer MSDepartment Of Biomedical Sciences, Cummings School Of Veterinary Medicine, Tufts University, Massachusetts, United States
Tel:+1 5088398788,
Fax:+1 5088398787
Email:Sawkat.anwer@tufts.edu
Abstract
This review summarizes our current understanding of the role of p38 MAPK in bile formation and more specifically in plasma membrane localization of MRP2. Bile formation is an energy-dependent process and requires vectorial transport of solutes from the sinusoidal space to the canaliculus. The coordinated function of transporters located at the sinusoidal and canalicular membranes results in the accumulation of solutes in the canalicular space providing the osmotic driving force for bile formation. Transport of organic anions by sinusoidal Na+-Taurocholate Cotransporting Polypeptide (NTCP) and canalicular Bile Salt Export Pump (BSEP) and Multidrug Resistance Protein 2 (MRP2) are primarily involved in bile formation. Cholestasis results when the ability of the liver to transport solutes into the canaliculus is compromised. These transporters undergo transcriptional and post-translational regulation. Transcriptional regulations are delayed effects of cholestasis that assure long term adjustments of transporter functions. On the other hand, post-translational regulations involve short-term rapid changes in Plasma Membrane (PM) localization of transporters permitting rapid changes in bile formation. P38 MAPK is one of the signaling pathways implicated in the regulation of PM localizations of BSEP and MRP2. Interestingly, p38 MAPK is involved in the insertion into as well as the retrieval of MRP2 from PM. A recent study provides evidence that the insertion into PM and the retrieval from PM of MRP2 are facilitated by p38α and p38β MAPKs, respectively. These p38MAPK isoforms are likely to be in turn regulated by various PKCs. The effect of p38 MAPK isoforms in bile formation and cholestasis should be taken into consideration when developing p38 MAPK inhibitors for inflammatory diseases to avoid liver toxicity.
INTRODUCTION
Transporters in bile formation
Transporter regulation: It is now well established that apart from genetic defects [15-18], regulation of these transporters at the level of transcriptional, translational and post-translational modifications may be altered by chemicals/disease processes. Indeed, cholestasis is associated with down-regulation of NTCP and MRP2 [19,20] with a relatively preserved expression of BSEP [19]. It is becoming evident that transcriptional regulation of hepatocellular transporters is primarily mediated via the nuclear receptor superfamily [3,21], while the post-translational regulation is mediated via classical second messengers. The transcriptional changes are delayed effects of cholestasis that assure long term adjustments of transporter functions. On the other hand, post-translational regulations involve short-term rapid changes in Plasma Membrane (PM) localization of transporters [1,5,6] and these permit rapid changes in bile formation. This review focusses on short-term regulations. The long term transcriptional regulations have recently been reviewed by others [3,21].
Short-term regulation: A transporter must be translocated to the appropriate membrane for it to transport its solute across that membrane. This is a complex regulated process requiring participations of various signaling molecules along with vesicles and cytoskeletons. A breakdown in this regulated process can lead to a decreased amount or an absence of a transport protein at its intended site resulting in decreased or no transport function and hence cholestasis. It is becoming evident that a translocation defect is an important event in cholestasis. Thus, experimental cholestasis induced by bile duct ligation, endotoxin, TLC and Estradiol-17beta-D-glucuronide (E-17G) is associated with decreased level of NTCP [22], MRP2 [20] and BSEP [23,24] in the plasma membrane. Certain other agents (cAMP, TC, TUDC) have been shown to stimulate translocation of these transporters to the membrane [12,25-29]. Acute cholestasis induced by TLC and E-17G is associated with the retrieval of BSEP and MRP2 from the canalicular membrane and these effects are reversed by cAMP [23,24,30]. TUDC has been shown to reverse TLC-induced retrieval of MRP2 [27,31]. These results highlight the importance of translocation defect in acute cholestasis. Thus, an understanding of the cellular mechanisms regulating transporter translocation and retrieval is necessary for medical management of patients with cholestasis.
A number of signaling pathways including Protein Kinase Cs (PKCs), p38 Mitogen Activated Protein Kinases (p38MAPKs), Rab proteins, Protein Phosphatases (PPs), actin binding proteins, ca2+ and Phosphoinosotide-3-Kinases (PI3Ks), [1,5,6,32,33] have been implicated in the regulation of PM localization of transporters (NTCP, BSEP and MRP2) by cAMP, TUDC, E17G and TLC [1,5,34,35]. In this review, we discuss the regulation by p38 MAPK as the role of p38 MAPK has not recently been reviewed.
P38 MAPK
Mammalian cells have four major types of MAPKs cascade and these include ERK1/2, JNKs, p38 MAPK and ERK5 cascades [36-40] (Figure 1).
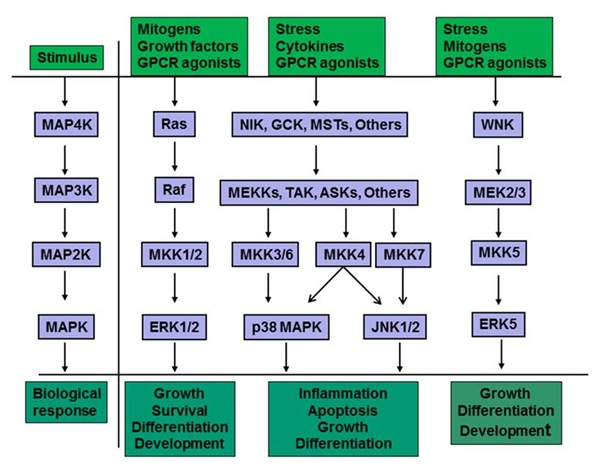 Figure 1: MAPK signaling cascades consist of a core of three sequentially activated protein kinases and these include MAPK kinase kinase (MAP3K), MAPK kinase (MAP2K) and MAPK [37-39,41]. In addition, there is an upstream MAPK kinase kinase (MAP4K) and a downstream MAPK activated protein kinase (MAPKAPK, not shown) in certain cells and for certain stimulations, but they are not always necessary for signaling through the cascades. Each cascade is initiated following extracellular stimulus. The extracellular stimuli include growth factors, mitogen, G-Protein Coupled Receptor (GPCR) agonist, stress and inflammatory cytokines. Activation of MAP4K typically involves phosphorylation by protein kinases activated by interaction of an agonist with its cell surface receptor. MAP3K, which is phosphorylated and activated by MAP4K, directly phosphorylates and activates MAP2K (also known as MKK or MEK). Activation of MAPK is then accomplished by dual phosphorylation of a conserved tripeptide (Thr-X-Tyr) motif in the conserved segment by MAP2K. MAP kinases include Extracellular signal-Regulated Kinase (ERK1/2), c-Jun amino-terminal Kinases (JNK1/2/3), p38 MAPK and ERK5. Activation of each MAPK leads to a diverse array of biological response.
Figure 1: MAPK signaling cascades consist of a core of three sequentially activated protein kinases and these include MAPK kinase kinase (MAP3K), MAPK kinase (MAP2K) and MAPK [37-39,41]. In addition, there is an upstream MAPK kinase kinase (MAP4K) and a downstream MAPK activated protein kinase (MAPKAPK, not shown) in certain cells and for certain stimulations, but they are not always necessary for signaling through the cascades. Each cascade is initiated following extracellular stimulus. The extracellular stimuli include growth factors, mitogen, G-Protein Coupled Receptor (GPCR) agonist, stress and inflammatory cytokines. Activation of MAP4K typically involves phosphorylation by protein kinases activated by interaction of an agonist with its cell surface receptor. MAP3K, which is phosphorylated and activated by MAP4K, directly phosphorylates and activates MAP2K (also known as MKK or MEK). Activation of MAPK is then accomplished by dual phosphorylation of a conserved tripeptide (Thr-X-Tyr) motif in the conserved segment by MAP2K. MAP kinases include Extracellular signal-Regulated Kinase (ERK1/2), c-Jun amino-terminal Kinases (JNK1/2/3), p38 MAPK and ERK5. Activation of each MAPK leads to a diverse array of biological response.
Each of these cascades consists of a core module of three tiers of protein kinases termed MAPK, MAP2K, and MAP3K. There are seven MAP2Ks (also known as MEK, MAP/ERK kinase, or MKK) that differentially activate different MAPKs by dual phosphorylation on Thr and Tyr. Thus, ERK1/2 is activated by MKK1/2, p38 MAPKs are activated by MKK3, MKK4, and MKK6, JNKs by MKK4/7 and ERK5 by MKK5. The MAPKs regulate members of a family of protein Ser/Thr kinases termed MAPK-Activated Protein Kinases (MAPKAPKs). The MAPKAPKs are related enzymes that respond to extracellular stimulation through direct MAPK-dependent activation loop phosphorylation and kinase activation [42]. The deactivation of MAPKs is achieved through dephosphorylation catalyzed by MAPK-specific Phosphatases (MKPs) including dual specific MAPK phosphatases [43,44]. MAPKs mediate/regulate diverse cellular functions including embryogenesis, apoptosis, immunity, proliferation, and differentiation by integrating signals from intra- and extracellular stimuli [41,45-47]. The p38 MAP kinase pathway shares many similarities with the other MAP kinase cascades, being associated with inflammation, cell growth, cell differentiation and cell death [48,49].
Activation stimuli: p38 MAPK is also known as Stress-Activated Protein Kinase (SAPK) since it is mostly activated by stresses such as ER stress, oxidative stress, metabolic stress and DNA damage, and by inflammatory cytokines [42,50-54]. The activation of p38 MAPK by a variety of stimuli (Figure 1) involves dual phosphorylation on Thr180 and Tyr182 by upstream MAPK kinases. While three upstream MAPK Kinases (MKKs), namely MKK3, MKK6 and MKK4, control the activation of p38 MAPK, MKK3 and MKK6 are considered the major activators of p38 MAPK [55-58]. A MAPKK-independent mechanism of p38 activation involves TAB1 (Transforming growth factor-beta-Activated protein Kinase 1 (TAK1)-binding protein 1) [59]. Interacting of TAB1 with p38 MAPK induces p38 autophosphorylation [60]. Inactivation of p38 MAPK involves dephosphorylation by phosphatases targeting threonine and tyrosine residues in the activation loop [61]. Phosphatases involved are Protein Phosphatase (PP) 2A add PP2C or Striatal Enriched Tyrosine Phosphatase (STEP) and Haemopoietic Tyrosine Phosphatase (HePTP) [49]. Inhibition of PP2A increases p38 activity by various stimuli [62-64]. In addition, microRNAs contribute to the homeostatic signaling of p38 MAPK pathway [40].
P38 MAPK isoforms: There are four known isoforms (α, β, γ and δ) of p38 MAPK with 60% homology and only α and β isoforms are expressed in livers [49,65,66]. Whereas p38α is ubiquitously expressed, p38β is mostly found in the brain, p38γ is predominantly expressed in skeletal muscle and p38δ gene expression is found in the lung, kidney, testis, pancreas and small intestine [49,67]. Activation of isoforms by upstream kinases differs based on stimuli and cell type [49,68,69]. In general, MKK6 can phosphorylate all four p38 MAPK family members, where as MKK3 activates p38α, p38γ and p38δ, but not p38β. In non-hepatic cells, MKK6 preferentially activates p38β MAPK [70] and MKK3 activates only p38α and p38? MAPKs [71]. Thus, MKK3 is expected to activate p38α and not p38β MAPK in the liver. In addition, p38α can also be phosphorylated by MKK4. Cyclic AMP has been shown to specifically activate p38α, but not p38β in adipocytes [72]. Selective activation of p38 isoforms has been reported in other cells [47,73,74]. Studies also suggest isoform specific effects of p38 MAPK. For example, p38α MAPK is involved in inflammation induced by proinflammatory cytokines, IL-1 and TNFα [75]. P38α and β MAPK have been shown to mediate cell cycle [67]. P38α but not p38β MAPK regulates chronic pain development [76]. P38α promotes apoptosis and p38β inhibits apoptosis [77,78]. P38δ synergistically regulates ERK pathway, whereas p38α medicates nuclear factor kappa B (NF?B) pathway for drug resistance [79].
Hepatic effects of p38 MAPK: Many hepatic functions are affected by p38MAPK. These functions include regulation of proliferation [80], protection against hypoxic injury [81], gluconeogenesis [82], enhancement hepatic ketogenesis [83], improvement of hepatic steatosis [84], inhibition on the progression of liver fibrosis [85], involvement in the pathogenesis and progression of intrahepatic cholestasis of pregnancy [86], bile acid synthesis [87] and anti-apoptotic effect of TUDC [88], bile acid-induced apoptosis [89,90], and biliary excretion of bile acids [91]. The p38 MAPK also regulates transporter translocation. For example, p38 MAPK is involved in GLUT4 translocation [92], EGF receptor endocytosis [93] and serotonin transporter trafficking [94]. In the liver, the p38 MAPK pathway is involved in the PM translocation/retrieval of MRP2 and BSEP [95,96]. Note that PM translocation of NTCP is not regulated by p38 MAPK [97,98]. Thus, the regulation of PM translocation/retrieval of MRP2 and BSEP by p38 MAPK in hepatocytes is discussed below.
HEPATOCELLULAR TRANSPORTER TRANSLOCATION AND P38 MAPK
Both choleretic (cAMP, TUDC, TC) and cholestatic agents (TLC, TDC and E17-G) have been employed to determine the cellular mechanisms regulating PM localization of BSEP and MRP2. These studies suggest a role for p38 MAPK in PM insertion/retrieval of BSEP and MRP2. TUDC-induced increases in bile acid secretion and BSEP translocation to the canalicular membrane require PI3K-independent activation of p38 MAPK [91,96]. Translocation of MRP2 to PM by cAMP is also mediated by p38 MAPK [99]. E-17G-induced endocytosis of BSEP and MRP2 is associated with decreased bile flow and biliary excretion of BSEP/MRP2 substrates and p38 inhibition prevents the internalization of BSEP/MRP2 [95]. P38 MAPK plays a role in oxidative stress-induced retrieval of BSEP and MRP2 [100]. TLC activates p38 MAPK [101] and decreases PM localization of MRP2 in hepatocytes [27,102]. These results suggest that p38 MAPK is involved in the insertion of BSEP/MRP2 to PM by choleretic agents as well as the retrieval of BSEP/MRP2 from PM by cholestatic agents. These paradoxical effects of p38 MAPK on retrieval as well as insertion of BSEP/MRP2 to PM led to the hypothesis that the effects of p38 MAPK on insertion and retrieval of these transporters may be mediated via different p38 MAPK isoforms. This hypothesis is based on findings that p38α MAPK and p38β MAPK have reciprocal effects on the expression of inducible nitric oxide synthase in renal mesangial cells [103].
There are limited studies on the role of p38 MAPK isoforms in the retrieval/insertion of hepatic transporters. One study showed that cAMP selectively activates p38α via activation of MKK3 and knockdown of p38α MAPK inhibited cAMP-induced insertion of MRP2 in a hepatic cell line [99]. These results were confirmed in studies with hepatocytes from MKK3 knockout mice [104]. In contrast to cAMP, TLC did not activate MKK3 or p38α MAPK in wild type mice hepatocytes and still decreased PM-MRP2 in MKK3 knockout hepatocytes. Additionally, TLC activated MKK6 in MKK3 knockout hepatocytes, and knockdown of p38β MAPK abrogated TLC-mediated decreases in PM-MRP2 in a hepatic cell line [104]. Taken together, these results suggest that activation of the MKK3/p38α MAPK pathway facilitates plasma membrane insertion of MRP2, whereas activation of the MKK6/p38β MAPK pathway mediates retrieval of PM-MRP2 by TLC (Figure 2). Whether insertion/retrieval of BSEP is also similarly regulated by P38 MAPK isoforms remains to be established.
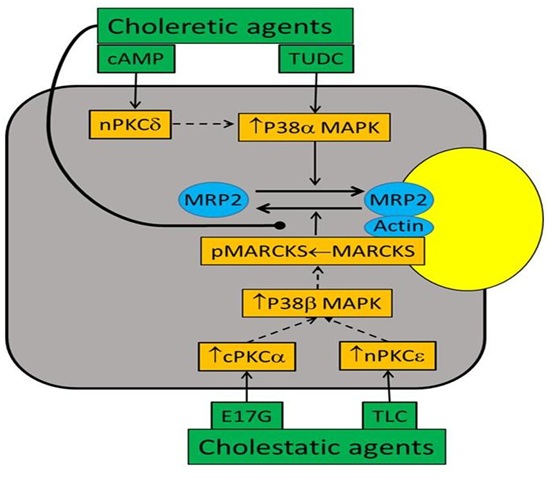 Figure 2: Regulation of PM MRP2 by p38 MAPK and PKCs. Not yet established but likely pathways are indicated by dotted lines. Choleretic agents, cAMP (acting via nPKCδ) and TUDC, increase PM-MRP2 by activating p38αMAPK. Cholestatic agents, E17G and TLC, decrease PM-MRP2 by activating cPKCα and nPKCε, respectively, followed by activation of p38βMAPK and MARCKS phosphorylation. Choleretic agents reverse PM-MRP2 retrieval by inhibiting MARCKS phosphorylation.
Figure 2: Regulation of PM MRP2 by p38 MAPK and PKCs. Not yet established but likely pathways are indicated by dotted lines. Choleretic agents, cAMP (acting via nPKCδ) and TUDC, increase PM-MRP2 by activating p38αMAPK. Cholestatic agents, E17G and TLC, decrease PM-MRP2 by activating cPKCα and nPKCε, respectively, followed by activation of p38βMAPK and MARCKS phosphorylation. Choleretic agents reverse PM-MRP2 retrieval by inhibiting MARCKS phosphorylation.
CROSSTALK BETWEEN P38 MAPK AND PKC SIGNALING PATHWAYS
Cellular mechanisms by which activation of p38 MAPK leads to alteration of PM localization of MRP2 may involve interactions with other signaling pathways. There are studies suggesting that Protein Kinase C (PKC) isoform also differentially regulate PM localization of BSEP and MRP2. PKC comprises a family of at least 12 isoforms [105], which include conventional (cPKCα, β, βI, βII and ?), novel (nPKCδ, ε, ? and ?), atypical (aPKC? and ?) isoforms and PKC?. cPKCα, nPKCδ, nPKCε, aPKC? and probably cPKCβII are present in hepatocytes [106-108]. For example, cPKCα mediates MRP2 and BSEP retrieval by E17G [109] and PMA [110], nPKCδ mediates cAMP-induced translocation of MRP2 to the plasma membrane [111], and nPKCε is responsible for the internalization of MRP2 by TLC [102]. Since PKCs activate p38MAPK in hepatocytes [95,112-114], it is likely that PKCs act upstream of p38 MAPK in hepatocytes (the regulation of PM localization of BSEP and MRP2). Indeed, results of several studies are consistent with this hypothesis. For example, hypoxia induced activation of p38 MAPK is blocked by PKC inhibitors in chicken hepatocytes [112]. Activation of p38 MAPK by PKCδ and PKCε has been reported in ischemic preconditioning in rat hepatocytes [115]. nPKCδ has been suggested to initiate p38 MAPK activation in butyrate-induced apoptosis in human colon adenoma cells [116]. One study suggests that cPKCs act upstream of p38 MAPK in E17G-induced retrieval of PM-BSEP and PM-MRP2 [95]. These studies raise the possibility that different PKCs may activate different p38MAPKs and this may explain isoform-specific effects of PKCs and p38MAPK on PM-MRP2. More specifically, activation of p38α MAPK by nPKCδ may facilitate MRP2 translocation to PM and activation of p38β MAPK by cPKCα and/or nPKCε may promote retrieval from PM (Figure 2). A preliminary study (unpublished) by authors showing that cAMP fails to activate p38α MAPK in PKCδ knockout hepatocytes is consistent with this hypothesis. However, further studies will be required to define whether PM-MRP2 localization involves isoform-specific effects of PKCs on p38α and p38β MAPKs in hepatocytes.
LIVER FUNCTIONS, INFLAMMATORY DISEASES AND P38 MAPK
The possibility of p38 MAPK as the therapeutic target for Rheumatoid Arthritis (RA) was raised with the finding in 1994 that a p38 inhibitor blocked Lipopolysaccharide (LPS)-induced TNFα and IL1β production by monocytes [75]. P38 MAPK inhibitor appeared to be a potential “wonder drug” and work began in earnest to synthesize and clinically evaluate novel inhibitors for inflammatory diseases [117-119]. These compounds were mainly competitive antagonists that blocked ATP binding to the kinase [120]. AP-1-dependent gene expression is p38 isoform specific in human breast cancer cells [121]. However, potency, lack of selectivity and toxicity limited their utility [122]. These compounds inhibited p38α and β but not the γ or δ isoforms [123]; at higher concentrations many other kinases were blocked [124]. While effective in preclinical models, a variety of toxicity problems, especially affecting the liver, interfered with clinical development [117]. Clinical trials with certain p38 MAPK inhibitors were discontinued because of liver toxicity [117,125], which is likely to be due to the inhibition of p38α MAPK. This is consistent with studies suggesting that deficiency of p38α in the liver increases the expression of chemokines to recruit more inflammatory cells [126], facilitates N-Nitrosodiethylamine (DEN) - induced hepatocellular carcinoma and increased proliferation [127,128]. In addition, p38α MAPK facilitates bile formation by translocating MRP2 to the PM. Recent studies suggest that MKK6 is a potential therapeutic target in RA [120,129]. Since MKK6 activates p38β but not p38α MAPK in hepatocytes and p38β MAPK induces cholestasis by retrieving MRP2 from the PM, MKK6 inhibitors may be less toxic to the liver. A better understanding of underlying signaling pathways of p38 MAPK should allow us to develop the drug target limiting hepatic toxicity and thereby improving the efficacy of p38 MAPK inhibitors in inflammatory diseases.
In summary, our understanding of cellular mechanisms underlying bile formation and cholestasis is steadily increasing. Recent studies suggest that p38α MAPK facilitates bile formation by inserting MRP2 into PM, while p38β MAPK mediates cholestasis by retrieving MRP2 from the PM. It appears that isoform specific effects of PKCs may be mediated via p38 MAPK isoforms. Since p38 MAPK is also involved in inflammation, development of drugs for inflammatory diseases by inhibiting p38 MAPK should take into account the effect of p38 MAPK isoforms in bile formation and cholestasis.
ACKNOWLEDGEMENT
The authors thank Dr. Webster for helpful discussion and Trena Haroutunian for secretarial assistance. The studies by authors referenced here were in part supported by a National Institutes of Health grant DK-33436.
REFERENCES
- Boyer JL (2013) Bile Formation and Secretion. Compr Physiol 3: 1035-1078.
- Maillette de Buy Wenniger L, Beuers U (2010) Bile salts and cholestasis. Dig Liver Dis 42: 409-418.
- Halilbasic E, Claudel T, Trauner M (2013) Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol 58: 155-168.
- Hofmann AF (2009) The enterohepatic circulation of bile acids in mammals: form and functions. Front Biosci (Landmark Ed) 14: 2584-2598.
- Anwer MS (2004) Cellular regulation of hepatic bile acid transport in health and cholestasis. Hepatology 39: 581-590.
- Crocenzi FA, Zucchetti AE, Boaglio AC, Barosso IR, Sanchez Pozzi EJ, et al. (2012) Localization status of hepatocellular transporters in cholestasis. Front Biosci (Landmark Ed) 17: 1201-1218.
- Wolkoff AW (2014) Organic Anion Uptake by Hepatocytes. Compr Physiol 4: 1715-1735.
- Stapelbroek JM, van Erpecum KJ, Klomp LW, Houwen RH (2010) Liver disease associated with canalicular transport defects: current and future therapies. J Hepatol 52: 258-271.
- Meier PJ, Stieger B (2000) Molecular Mechanisms in Bile Formation. News Physiol Sci 15: 89-93.
- Wolkoff AW, Cohen DE (2003) Bile acid regulation of hepatic physiology: I. Hepatocyte transport of bile acids. Am J Physiol Gastrointest Liver Physiol 284: 175-179.
- Trauner M, Boyer JL (2003) Bile salt transporters: molecular characterization, function, and regulation. Physiol Rev 83: 633-671.
- Marinelli RA, Tietz PS, LaRusso NF (2005) Regulated vesicle trafficking of membrane transporters in hepatic epithelia. J Hepatol 42: 592-603.
- Anwer MS (2004) Mechanisms of bile formation and cholestasis. Principles of Medical Biology 15: 81-118.
- Jemnitz K, Heredi-Szabo K, Janossy J, Ioja E, Vereczkey L, et al (2010) ABCC2/Abcc2: a multispecific transporter with dominant excretory functions. Drug Metab Rev 42: 402-436.
- Paulusma CC, Kool M, Bosma PJ, Scheffer GL, ter Borg F, et al. (1997) A mutation in the human canalicular multispecific organic anion transporter gene causes the Dubin-Johnson syndrome. Hepatology 25: 1539-1542.
- Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, et al. (1998) A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet 20: 233-238.
- De Vree JM, Jacquemin E, Sturm E, Cresteil D, Bosma PJ, et al. Mutations in the MDR3 gene cause progressive familial intrahepatic cholestasis. Proc Natl Acad Sci USA 95: 282-287.
- Wang L, Soroka CJ, Boyer JL (2002) The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J Clin Invest 10: 965-972.
- Lee JM, Trauner M, Soroka CJ, Stieger B, Meier PJ, et al (2000) Expression of the bile salt export pump is maintained after chronic cholestasis in the rat. Gastroenterology 118: 163-172.
- Trauner M, Arrese M, Soroka CJ, Ananthanarayanan M, Koeppel TA, et al. (1997) The rat canalicular conjugate export pump (Mrp2) is down-regulated in intrahepatic and obstructive cholestasis. Gastroenterology 113: 255-264.
- Chiang JY (2013) Bile acid metabolism and signaling. Compr Physiol 3: 1191-1212.
- Gartung C, Ananthanarayanan M, Rahman MA, Schuele S, Nundy S, et al. (1996) Down-regulation of expression and function of the rat liver Na+/bile acid cotransporter in extrahepatic cholestasis. Gastroenterology 110: 199-209.
- Crocenzi FA, Mottino AD, Cao J, Veggi LM, Pozzi EJ, Vore M, et al. (2003) Estradiol-17beta-D-glucuronide induces endocytic internalization of Bsep in rats. Am J Physiol Gastrointest Liver Physiol 285: 449-459.
- Crocenzi FA, Mottino AD, Sánchez Pozzi EJ, Pellegrino JM, Rodríguez Garay EA, et al. (2003) Impaired localisation and transport function of canalicular Bsep in taurolithocholate induced cholestasis in the rat. Gut 52: 1170-1177.
- Roelofsen H, Soroka CJ, Keppler D, Boyer JL (1998) Cyclic AMP stimulates sorting of the canalicular organic anion transporter (Mrp2/cMoat) to the apical domain in hepatocyte couplets. J Cell Sci 111: 1137-1145.
- Gatmaitan ZC, Nies AT, Arias IM (1997) Regulation and translocation of ATP-dependent apical membrane proteins in rat liver. Am J Physiol 272: 1041-1049.
- Beuers U, Bilzer M, Chittattu A, Kullak-Ublick GA, Keppler D, et al. (2001) Tauroursodeoxycholic acid inserts the apical conjugate export pump, Mrp2, into canalicular membranes and stimulates organic anion secretion by protein kinase C-dependent mechanisms in cholestatic rat liver. Hepatology 33: 1206-1216.
- Kipp H, Pichetshote N, Arias IM (2001) Transporters on demand: intrahepatic pools of canalicular ATP binding cassette transporters in rat liver. J Biol Chem 276: 7218-7224.
- Dranoff JA, McClure M, Burgstahler AD, Denson LA, Crawford AR, et al. (1999) Short-term regulation of bile acid uptake by microfilament-dependent translocation of rat ntcp to the plasma membrane. Hepatology 30: 223-229.
- Mottino AD, Cao J, Veggi LM, Crocenzi F, Roma MG, et al. (2002) Altered localization and activity of canalicular Mrp2 in estradiol-17beta-D-glucuronide-induced cholestasis. Hepatology 35: 1409-1419.
- Beuers U, Denk GU, Soroka CJ, Wimmer R, Rust C, et al. (2003) Taurolithocholic acid exerts cholestatic effects via phosphatidylinositol-3 kinase-dependent mechanisms in perfused rat livers and rat hepatocyte couplets. J Biol Chem 278: 17810-17818. (2012) Localization status of hepatocellular transporters in cholestasis. Front Biosci (Landmark Ed) 17: 1201-1218.
- Anwer MS, Webster CRL (2004) Signal Transduction in bile formation and cholestasis. In: Trauner M, Jansen PLM (eds.). Molecular Pathogenesis of Cholestasis. Landes Bioscience, Austin, Texas, USA.
- Anwer MS (2014) Role of protein kinase C isoforms in bile formation and cholestasis. Hepatology 60: 1090-1097.
- Nies AT, Keppler D (2007) The apical conjugate efflux pump ABCC2 (MRP2). Pflugers Arch 453: 643-659.
- Gerk PM, Vore M (2002) Regulation of expression of the multidrug resistance-associated protein 2 (MRP2) and its role in drug disposition. J Pharmacol Exp Ther 302: 407-415.
- Morrison DK (2012) MAP kinase pathways. Cold Spring Harb Perspect Biol 4: 011254.
- Avruch J (2007) MAP kinase pathways: the first twenty years. Biochim Biophys Acta 1773: 1150-1160.
- Krishna M, Narang H (2008) The complexity of mitogen-activated protein kinases (MAPKs) made simple. Cell Mol Life Sci 65: 3525-3544.
- Keshet Y, Seger R (2010) The MAP kinase signaling cascades: a system of hundreds of components regulates a diverse array of physiological functions. Methods Mol Biol 661: 3-38.
- Corre I, Paris F, Huot J (2017) The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget 8: 55684-55714.
- Raman M, Chen W, Cobb MH (2007) Differential regulation and properties of MAPKs. Oncogene 26: 3100-3112.
- Cargnello M, Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75: 50-83.
- Kondoh K, Nishida E (2007) Regulation of MAP kinases by MAP kinase phosphatases. Biochim Biophys Acta 1773: 1227-1237.
- Caunt CJ, Keyse SM (2013) Dual-specificity MAP kinase phosphatases (MKPs): shaping the outcome of MAP kinase signalling. FEBS J 280: 489-504.
- Cook R, Wu CC, Kang YJ, Han J (2007) The role of the p38 pathway in adaptive immunity. Cellular & Molecular Immunology 4: 253-259.
- Han J, Sun P (2007) The pathways to tumor suppression via route p38. Trends Biochem Sci 32: 364-371.
- Kaminska B (2005) MAPK signalling pathways as molecular targets for anti-inflammatory therapy--from molecular mechanisms to therapeutic benefits. Biochim Biophys Acta 1754: 253-262.
- Sun Y, Liu WZ, Liu T, Feng X, Yang N, et al. (2015) Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J Recept Signal Transduct Res 35: 600-604.
- Cuadrado A, Nebreda AR (2010) Mechanisms and functions of p38 MAPK signalling. Biochem J 429: 403-417.
- Roux PP, Blenis J (2004) ERK and p38 MAPK-activated protein kinases: a family of protein kinases with diverse biological functions. Microbiol Mol Biol Rev 68: 320-344.
- Kyriakis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81: 807-869.
- Choy EH, Panayi GS (2001) Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med 344: 907-916.
- Wang J, Chen L, Ko CI, Zhang L, Puga A, et al. (2012) Distinct signaling properties of mitogen-activated protein kinase kinases 4 (MKK4) and 7 (MKK7) in embryonic stem cell (ESC) differentiation. J Biol Chem 287: 2787-2797.
- Meng F, Yamagiwa Y, Taffetani S, Han J, Patel T (2005) IL-6 activates serum and glucocorticoid kinase via p38alpha mitogen-activated protein kinase pathway. Am J Physiol Cell Physiol 289: 971-981.
- Koul HK, Pal M, Koul S (2013) Role of p38 MAP Kinase Signal Transduction in Solid Tumors. Genes Cancer 4: 342-359.
- Derijard B, Raingeaud J, Barrett T, Wu IH, Han J, et al. (1995) Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 267: 682-685.
- Raingeaud J, Whitmarsh AJ, Barrett T, Derijard B, Davis RJ (1996) MKK3- and MKK6-regulated gene expression is mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Mol Cell Biol 16: 1247-1255.
- Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, et al. (2003) Mechanism of p38 MAP kinase activation in vivo. Genes Dev 17: 1969-1978.
- Ge B, Gram H, Di PF, Huang B, New L, et al. (2002) MAPKK-independent activation of p38alpha mediated by TAB1-dependent autophosphorylation of p38alpha. Science 295: 1291-1294.
- DeNicola GF, Martin ED, Chaikuad A, Bassi R, Clark J, et al. (2013) Mechanism and consequence of the autoactivation of p38alpha mitogen-activated protein kinase promoted by TAB1. Nat Struct Mol Biol 20: 1182-1190.
- Risco A, Cuenda A (2012) New Insights into the p38γ and p38δ MAPK Pathways. J Signal Transduct 2012: 520289.
- Boudreau RT, Conrad DM, Hoskin DW (2007) Apoptosis induced by protein phosphatase 2A (PP2A) inhibition in T leukemia cells is negatively regulated by PP2A-associated p38 mitogen-activated protein kinase. Cell Signal 19: 139-151.
- Lin SP, Lee YT, Wang JY, Miller SA, Chiou SH, et al. (2012) Survival of cancer stem cells under hypoxia and serum depletion via decrease in PP2A activity and activation of p38-MAPKAPK2-Hsp27. PLoS One 7: 49605.
- Law AH, Tam AH, Lee DC, Lau AS (2013) A role for protein phosphatase 2A in regulating p38 mitogen activated protein kinase activation and tumor necrosis factor-alpha expression during influenza virus infection. Int J Mol Sci 14: 7327-7340.
- Jiang Y, Gram H, Zhao M, New L, Gu J, et al. (1997) Characterization of the structure and function of the fourth member of p38 group mitogen-activated protein kinases, p38delta. J Biol Chem 272: 30122-30128.
- Wang H, Xu Q, Xiao F, Jiang Y, Wu Z (2008) Involvement of the p38 mitogen-activated protein kinase alpha, beta, and gamma isoforms in myogenic differentiation. Mol Biol Cell 19: 1519-1528.
- Cuenda A, Rousseau S (2007) p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta 1773: 1358-1375.
- Zucchetti AE, Barosso IR, Boaglio AC, Luquita MG, Roma MG, et al. (2013) Hormonal modulation of hepatic cAMP prevents estradiol 17β-D-glucuronide-induced cholestasis in perfused rat liver. Dig Dis Sci 58: 1602-1614.
- Alonso G, Ambrosino C, Jones M, Nebreda AR (2000) Differential activation of p38 mitogen-activated protein kinase isoforms depending on signal strength. J Biol Chem 275: 40641-40648.
- Jiang Y, Chen C, Li Z, Guo W, Gegner JA, et al. (1996) Characterization of the structure and function of a new mitogen-activated protein kinase (p38beta). J Biol Chem 271: 17920-17926.
- Enslen H, Raingeaud J, Davis RJ (1998) Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem 273: 1741-1748.
- Robidoux J, Cao W, Quan H, Daniel KW, Moukdar F, et al. (2005) Selective activation of mitogen-activated protein (MAP) kinase kinase 3 and p38alpha MAP kinase is essential for cyclic AMP-dependent UCP1 expression in adipocytes. Mol Cell Biol 25: 5466-5479.
- Wang L, Ma R, Flavell RA, Choi ME (2002) Requirement of mitogen-activated protein kinase kinase 3 (MKK3) for activation of p38alpha and p38delta MAPK isoforms by TGF-beta 1 in murine mesangial cells. J Biol Chem 277: 47257-47262.
- Korb A, Tohidast-Akrad M, Cetin E, Axmann R, Smolen J, et al. (2006) Differential tissue expression and activation of p38 MAPK alpha, beta, gamma, and delta isoforms in rheumatoid arthritis. Arthritis Rheum 54: 2745-2756.
- Lee JC, Laydon JT, McDonnell PC, Gallagher TF, Kumar S, et al. (1994) A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature 372: 739-746.
- Luo X, Fitzsimmons B, Mohan A, Zhang L, Terrando N, et al. (2017) Intrathecal administration of antisense oligonucleotide against p38α but not p38β MAP kinase isoform reduces neuropathic and postoperative pain and TLR4-induced pain in male mice. Brain Behav Immun 72: 34-44.
- Kaiser RA, Lyons JM, Duffy JY, Wagner CJ, McLean KM, et al. (2005) Inhibition of p38 reduces myocardial infarction injury in the mouse but not pig after ischemia-reperfusion. Am J Physiol Heart Circ Physiol 289: 2747-2751.
- Nemoto S, Xiang J, Huang S, Lin A (1998) Induction of apoptosis by SB202190 through inhibition of p38beta mitogen-activated protein kinase. J Biol Chem 273: 16415-16420.
- Peng H, Peng T, Wen J, Engler DA, Matsunami RK, et al. (2014) Characterization of p38 MAPK isoforms for drug resistance study using systems biology approach. Bioinformatics 30: 1899-1907.
- Awad MM, Enslen H, Boylan JM, Davis RJ, Gruppuso PA (2000) Growth regulation via p38 mitogen-activated protein kinase in developing liver. J Biol Chem 275: 38716-38721.
- Carini R, Alchera E, Baldanzi G, Piranda D, Splendore R, et al. (2007) Role of p38 map kinase in glycine-induced hepatocyte resistance to hypoxic injury. J Hepatol 46: 692-699.
- Cao W, Collins QF, Becker TC, Robidoux J, Lupo EG, et al. (2005) p38 Mitogen-activated protein kinase plays a stimulatory role in hepatic gluconeogenesis. J Biol Chem 280: 42731-42737.
- Shi L, Zhao D, Hou C, Peng Y, Liu J, et al. (2017) Early interleukin-6 enhances hepatic ketogenesis in APPSWE/PSEN1dE9 mice via 3-hydroxy-3-methylglutary-CoA synthase 2 signaling activation by p38/nuclear factor kB p65. Neurobiol Aging 56: 115-126.
- Ou HY, Wu HT, Lu FH, Su YC, Hung HC, et al. (2014) Activation of free fatty acid receptor 1 improves hepatic steatosis through a p38-dependent pathway. J Mol Endocrinol 53: 165-174.
- Franca MER, Rocha SWS, Oliveira WH, Santos LA, de Oliveira AGV, et al. (2018) Diethylcarbamazine attenuates the expression of pro-fibrogenic markers and hepatic stellate cells activation in carbon tetrachloride-induced liver fibrosis. Inflammopharmacology 26: 599-609.
- Wu W-B, Menon R, Xu Y-Y, Zhao J-R, Wang Y-L, et al. (2016) Downregulation of peroxiredoxin-3 by hydrophobic bile acid induces mitochondrial dysfunction and cellular senescence in human trophoblasts. Sci Rep 6: 38946.
- 8Xu Z, Tavares-Sanchez OL, Li Q, Fernando J, Rodriguez CM, et al. (2007) Activation of bile acid biosynthesis by the p38 mitogen-activated protein kinase (MAPK): hepatocyte nuclear factor-4alpha phosphorylation by the p38 MAPK is required for cholesterol 7alpha-hydroxylase expression. J Biol Chem 282: 24607-24614.
- Schoemaker MH, Conde de la Rosa L, Buist-Homan M, Vrenken TE, Havinga R, et al. (2004) Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 39: 1563-1573.
- Grambihler A, Higuchi H, Bronk SF, Gores GJ (2003) cFLIP-L inhibits p38 MAPK activation: an additional anti-apoptotic mechanism in bile acid-mediated apoptosis. J Biol Chem 278: 26831-26837.
- Zhang T, Zhao C, Luo L, Xiang J, Cheng J, et al. (2014) High concentraction of taurocholic acid induced apoptosis in HTR-8/SVneo cells via overexpression of ERp29 and activation of p38. Placenta 35: 496-500.
- Kubitz R, Sütfels G, Kühlkamp T, Kölling R, Häussinger D (2004) Trafficking of the bile salt export pump from the Golgi to the canalicular membrane is regulated by the p38 MAP kinase. Gastroenterology 126: 541-553.
- Somwar R, Kim DY, Sweeney G, Huang C, Niu W, et al. (2001) GLUT4 translocation precedes the stimulation of glucose uptake by insulin in muscle cells: potential activation of GLUT4 via p38 mitogen-activated protein kinase. Biochem J 359: 639-649.
- Tomas A, Jones S, Vaughan SO, Hochhauser D, Futter CE (2017) Stress-specific p38 MAPK activation is sufficient to drive EGFR endocytosis but not its nuclear translocation. J Cell Sci 130: 2481-2490.
- Samuvel DJ, Jayanthi LD, Bhat NR, Ramamoorthy S (2005) A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: evidence for distinct cellular mechanisms involved in transporter surface expression. J Neurosci 25: 29-41.
- Boaglio AC, Zucchetti AE, Toledo FD, Barosso IR, Sánchez Pozzi EJ, et al. (2012) ERK1/2 and p38 MAPKs Are Complementarily Involved In Estradiol 17ß-d-Glucuronide-Induced Cholestasis: Crosstalk with cPKC and PI3K. PLoS One 7: 0049255.
- Kurz AK, Graf D, Schmitt M, Dahl SV, Häussinger D (2001) Tauroursodesoxycholate-induced choleresis involves p38(MAPK) activation and translocation of the bile salt export pump in rats. Gastroenterology 121: 407-419.
- Mühlfeld S, Domanova O, Berlage T, Stross C, Helmer A, et al. (2012) Short-term feedback regulation of bile salt uptake by bile salts in rodent liver. Hepatology 56: 2387-2397.
- Webster CR, Srinivasulu U, Ananthanarayanan M, Suchy FJ, Anwer MS (2002) Protein kinase B/Akt mediates cAMP- and cell swelling-stimulated Na+/taurocholate cotransport and Ntcp translocation. J Biol Chem 277: 28578-28583.
- Schonhoff CM, Webster CR, Anwer MS (2010) Cyclic AMP stimulates Mrp2 translocation by activating p38{alpha} MAPK in hepatic cells. Am J Physiol Gastrointest Liver Physiol 298: G667-G674.
- Toledo FD, Basiglio CL, Barosso IR, Boaglio AC, Zucchetti AE, et al. (2017) Mitogen-activated protein kinases are involved in hepatocanalicular dysfunction and cholestasis induced by oxidative stress. Arch Toxicol 91: 2391-2403.
- Graf D, Reinehr R, Kurz AK, Fischer R, Haussinger D (2013) Inhibition of taurolithocholate 3-sulfate-induced apoptosis by cyclic AMP in rat hepatocytes involves protein kinase A-dependent and -independent mechanisms. Arch Biochem Biophys 415: 34-42.
- Schonhoff CM, Webster CR, Anwer MS (2003) Taurolithocholate-induced MRP2 retrieval involves MARCKS phosphorylation by protein kinase C? in HUH-NTCP Cells. Hepatology 58: 284-292.
- Lui P, Zeng C, Acton S, Cok S, Sexton A, et al. (2004) Effects of p38MAPK isoforms on renal mesangial cell inducible nitric oxide synthase expression. Am J Physiol Cell Physiol 286: 145-152.
- Ramasamy U, Anwer MS, Schonhoff CM (2013) Cysteine 96 of Ntcp is responsible for NO-mediated inhibition of taurocholate uptake. Am J Physiol Gastrointest Liver Physiol 305: 513-519.
- Newton AC (2003) Regulation of the ABC kinases by phosphorylation: protein kinase C as a paradigm. Biochem J 370: 361-371.
- Beuers U, Probst I, Soroka C, Boyer JL, Kullak-Ublick GA, et al. (1999) Modulation of protein kinase C by taurolithocholic acid in isolated rat hepatocytes. Hepatology 29: 477-482.
- Jones BA, Rao YP, Stravitz RT, Gores GJ (1997) Bile salt-induced apoptosis of hepatocytes involves activation of protein kinase C. Am J Physiol 272: 1109-1115.
- Stravitz RT, Rao YP, Vlahcevic ZR, Gurley EC, Jarvis WD, et al. (1996) Hepatocellular protein kinase C activation by bile acids: implications for regulation of cholesterol 7 alpha-hydroxylase. Am J Physiol 271: 293-303.
- Crocenzi FA, Sanchez Pozzi EJ, Ruiz ML, Zucchetti AE, Roma MG, et al. (2008) Ca(2+)-dependent protein kinase C isoforms are critical to estradiol 17beta-D-glucuronide-induced cholestasis in the rat. Hepatology 48: 1885-1895.
- Kubitz R, Huth C, Schmitt M, Horbach A, Kullak-Ublick G, et al. (2001) Protein kinase C-dependent distribution of the multidrug resistance protein 2 from the canalicular to the basolateral membrane in human HepG2 cells. Hepatology 34: 340-350.
- Park SW, Schonhoff CM, Webster CR, Anwer MS (2012) Protein kinase Cdelta differentially regulates cAMP-dependent translocation of NTCP and MRP2 to the plasma membrane. Am J Physiol Gastrointest Liver Physiol 303: 657-665.
- Lee SH, Lee MY, Lee JH, Han HJ (2008) A potential mechanism for short time exposure to hypoxia-induced DNA synthesis in primary cultured chicken hepatocytes: Correlation between Ca(2+)/PKC/MAPKs and PI3K/Akt/mTOR. J Cell Biochem 104: 1598-1611.
- Mei Y, Thevananther S (2011) Endothelial nitric oxide synthase is a key mediator of hepatocyte proliferation in response to partial hepatectomy in mice. Hepatology 54: 1777-1789.
- Carini R, De Cesaris MG, Splendore R, Vay D, Domenicotti C, et al. (2001) Signal pathway involved in the development of hypoxic preconditioning in rat hepatocytes. Hepatology 33: 131-139.
- Calle R, Ganesan S, Smallwood JI, Rasmussen H (1992) Glucose-induced phosphorylation of myristoylated alanine-rich C kinase substrate (MARCKS) in isolated rat pancreatic islets. J Biol Chem 267: 18723-18727.
- McMillan L, Butcher SK, Pongracz J, Lord JM (2003) Opposing effects of butyrate and bile acids on apoptosis of human colon adenoma cells: differential activation of PKC and MAP kinases. Br J Cancer 88: 748-753.
- Hammaker D, Firestein GS (2010) “Go upstream, young man”: lessons learned from the p38 saga. Ann Rheum Dis 69: 77-82.
- Mayer RJ, Callahan JF (2006) p38 MAP kinase inhibitors: A future therapy for inflammatory diseases. Drug Discovery Today: Therapeutic Strategies 3: 49-54.
- Yong HY, Koh MS, Moon A (2009) The p38 MAPK inhibitors for the treatment of inflammatory diseases and cancer. Expert Opin Investig Drugs 18: 1893-1905.
- Wagner G, Laufer S (2006) Small molecular anti-cytokine agents. Med Res Rev 26: 1-62.
- Pramanik R, Qi X, Borowicz S, Choubey D, Schultz RM, et al. (2003) p38 isoforms have opposite effects on AP-1-dependent transcription through regulation of c-Jun. The determinant roles of the isoforms in the p38 MAPK signal specificity. J Biol Chem 278: 4831-4839.
- Kumar S, Boehm J, Lee JC (2003) p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov 2: 717-726.
- Lee JC, Kumar S, Griswold DE, Underwood DC, Votta BJ, et al. (2000) Inhibition of p38 MAP kinase as a therapeutic strategy. Immunopharmacology 47: 185-201.
- Fabian MA, Biggs WH 3rd, Treiber DK, Atteridge CE, Azimioara MD, et al. (2005) A small molecule-kinase interaction map for clinical kinase inhibitors. Nat Biotechnol 23: 329-336.
- Dambach DM (2005) Potential adverse effects associated with inhibition of p38alpha/beta MAP kinases. Curr Top Med Chem 5: 929-939.
- Kang YJ, Bang BR, Otsuka M, Otsu K (2015) Tissue-Specific Regulation of p38α-Mediated Inflammation in Con A-Induced Acute Liver Damage. J Immunol 194: 4759-4766.
- Hui L, Bakiri L, Mairhorfer A, Schweifer N, Haslinger C, et al. (2007) p38alpha suppresses normal and cancer cell proliferation by antagonizing the JNK-c-Jun pathway. Nat Genet 39: 741-749.
- Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, et al. (2008) Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell 14: 156-165.
- Hammaker D, Boyle DL, Topolewski K, Firestein GS (2014) Differential regulation of anti-inflammatory genes by p38 MAP kinase and MAP kinase kinase 6. J Inflamm (Lond) 11: 14.
Citation: Park SW, Anwer MS (2018) Role of P38 MAPK in Bile Formation and Cholestasis. J Gastroenterol Hepatology Res 3: 022.
Copyright: © 2018 Park SW, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.