Sodium Transport and Alveolar Fluid Clearance in Acute Lung Injury
*Corresponding Author(s):
Bo LiDepartment Of Human Anatomy, College Of Basic Medical Sciences, Jilin University, Changchun, China
Email:libosf@gmail.com
Abstract
Acute Lung Injury (ALI) and Acute Respiratory Distress Syndrome (ARDS) are characterised by pulmonary oedema results from increased vascular permeability. The resolution of pulmonary oedema and ALI depends upon intact Alveolar Fluid Clearance (AFC). Sodium transport Across Alveolar Epithelial Cells (AECs) leads to osmotic alveolar water transport and plays a dominant role in AFC. Sodium transports via apical sodium channels, mainly the Epithelial Sodium Channel (ENaC) and basolateral Sodium-Potassium Adenosine Triphosphatase (Na+, K+-ATPase) in the AEC membrane. In ALI/ARDS, the imbalance of oxygen, Reactive Nitrogen and Oxygen Species (RNS and ROS, respectively), and Tumour Necrosis factor-α (TNF-α) lead to a decrease in AFC, due in part to the down regulation of ENaC and Na+, K+-ATPase in the alveolar epithelium.
In ALI, hypoxia inhibits ENaC and Na+, K+-ATPase activity through different mechanisms. The definite mechanism of ENaC and Na+, K+-ATPase activity regulation by RNS and ROS is unclear. TNF-α, and its lectin-like domain (designated TIP) differentially impact sodium transport across the alveolar epithelium.
In this review, we will discuss the regulatory mechanisms of alveolar sodium transport and AFC for the development of effective therapeutic strategies for ALI/ARDS patients.
Keywords
ABBREVIATIONS
AEC: Alveolar Epithelial Cell; AFC: Alveolar Fluid Clearance ; ALI: Acute Lung Injury; AMPK: Adenosine Monophosphate-activated Protein Kinase; AP-2: Adaptor Protein-2; ARDS: Acute Respiratory Distress Syndrome; AT I cell: Alveolar Type I cell; AT II cell: Alveolar Type II cell; ATP: Adenosine Triphosphate; cAMP: cyclic AMP; CFTR: Cystic Fibrosis Transmembrane conductance Regulator; cldn: Claudin; Cl-: Chlorine; CNG cation channel: Cyclic Nucleotide-Gated cation channel; CRAC channel: Calcium Release-Activated Calcium channel; DEX: dexamethasone; ENaC: Epithelial Sodium Channel; FDLE cell: Foetal Distal Lung Epithelial Cell; GC: Glucocorticoid; GR: GC Receptor; HSC: Highly Selective Cation channel; iNOS: inducible NO Synthase; mROS: mitochondrial Reactive Oxygen Species; NO: Nitric Oxide; Na+: Sodium; Na+, K+-ATPase: Sodium-Potassium Adenosine Triphosphatase; NSC: Nonselective Cation channel; PLC: Phospholipase C; PKCζ: Protein Kinase C; PKA: Protein Kinase A; RNS: Reactive Nitrogen Species; ROS: Reactive Oxygen Species; SGK1: Serum and Glucocorticoid-inducible Kinase 1; TJ: Tight Junction; TNF-α: Tumour Necrosis factor-α; ZO-1: Zonula Occludens-1; ZO-2: Zonula Occludens-2; α-ENaC: ENaC α-subunit; α1-Na+, K+-ATPase: α1-isoform of Na+, K+-ATPase; ß2AR: ß2-Adrenergic Receptor
INTRODUCTION
Alveolar sodium and fluid transport
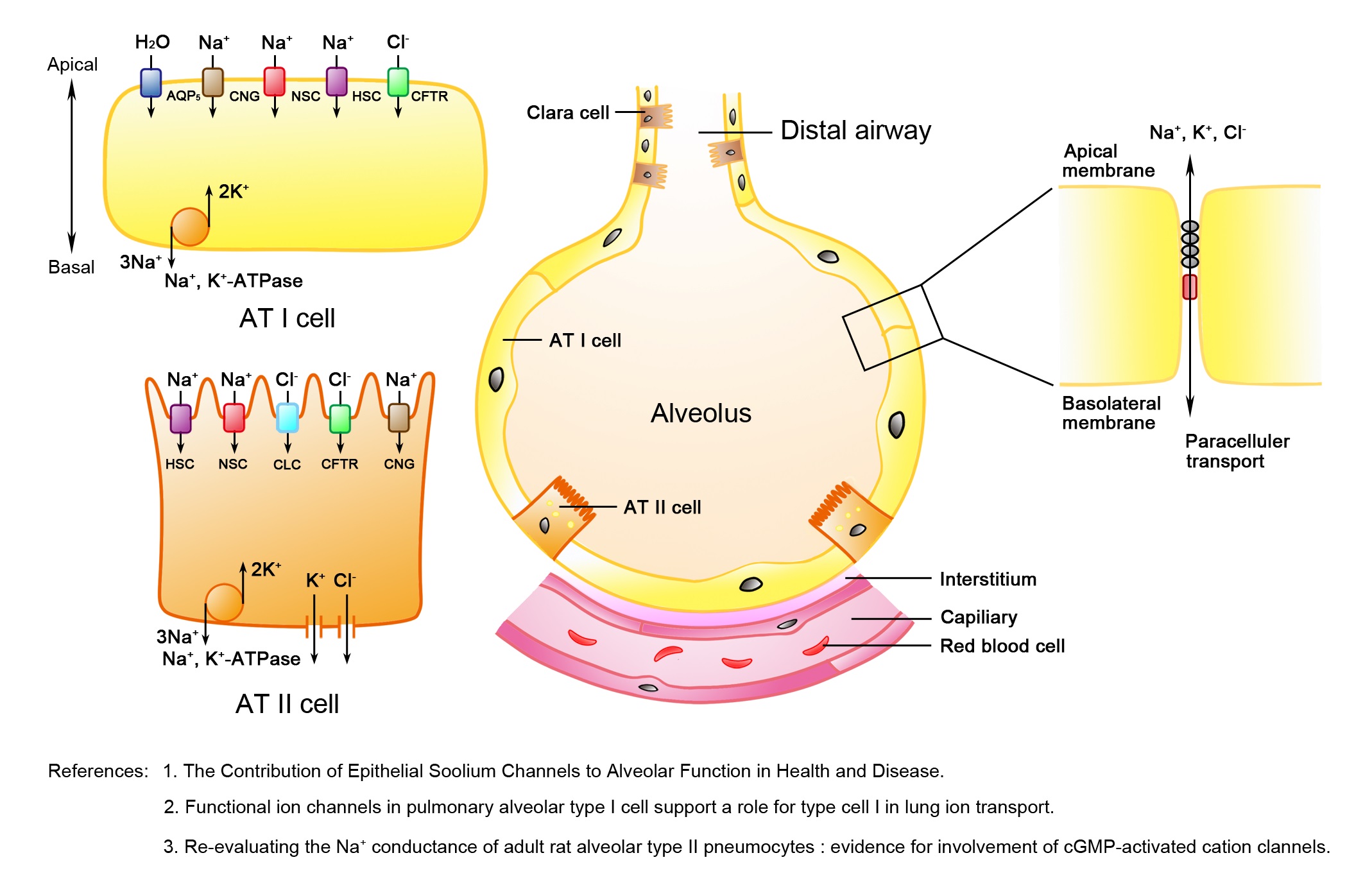 Figure 1: Schematic diagram of the distal pulmonary epithelium.
Figure 1: Schematic diagram of the distal pulmonary epithelium.The alveolar epithelium is composed of squamous Alveolar Type I (AT I) and cuboidal Alveolar Type II (AT II) cells. Both AT I and AT II cells contain two forms of amiloride-sensitive epithelial sodium channels, including the Highly Selective Cation channel (HSC) and Nonselective Cation channel (NSC) as well as the Cyclic Nucleotide-Gated (CNG) cation channel and sodium-potassium adenosine triphosphatase (Na+, K+-ATPase), which are involved in alveolar transepithelial sodium transport. In addition, AT I cells have aquaporin 5, which contributes to either water or gas exchange. AT II cells have the Cystic Fibrosis Transmembrane conductance Regulator (CFTR) and Chlorine (Cl-) channels, members of the CLC family of proteins, which mediate apical Cl- transport. The tight junctions (a chain in grey between Alveolar Epithelial Cells (AECs)) and adherens junctions (in red between AECs) between adjacent alveolar epithelial cells provide a physical barrier from paracellular solute transport [4,6,7].
Intercellular junctions between adjacent alveolar epithelial cells, such as Tight Junctions (TJs) and adherens junctions, provide a physical barrier for paracellular solute transport. TJ plays a more notable role in regulating paracellular solute transport. The proteins that compose the core TJ protein complex, including tetraspan transmembrane Claudin (cldn), occludin, and cytoplasmic scaffold proteins Zonula Occludens-1 (ZO-1) and ZO-2 [8], intersect one another in a coordinated manner and create junctional proteins strands, which surround the cells like a ring and divide the epithelium into apical and basolateral surfaces. Other proteins, such as cytoskeletal protein actin, are also known components of the TJ. Of these TJ proteins, recent studies have focused more on the cldn family because TJ regulation of paracellular ion transport is directly mediated by the cldn family, which includes nearly two dozen cldns [8]. The distribution of cldns is tissue-specific. In AECs (Figure 2), cldn-18.1 is abundantly expressed by AT I cells, cldn-3 is predominantly expressed by AT II cells, and cldn-4 is subdominantly expressed by AT I and AT II cells [9]. The function of these three claudins in the alveolar epithelium, especially cldn-3 and cldn-18.1, has not been fully explored. Cldn-4 is specifically upregulated in ALI [10] and increased cldn-4 expression may related to the resolution of pulmonary oedema [9]. Elevated cldn-4 expression can increase alveolar epithelial transepithelial electrical resistance in primary rat AECs [10], while inhibiting cldn-4 function has been shown to lead to decreased transepithelial electrical resistance and air space fluid clearance, thus impacting paracellular transport, AFC and sensitivity to pulmonary oedema in a ventilator-induced lung injury model [11]. Changes in cldn-18 expression in ALI also can be seen. However, the regulation of cldn-4 and cldn-18 is in opposite directions. In experimental bleomycin-induced lung injury, the level of mRNAs encoding tight junction proteins, especially claudin-18, were demonstrated to be down regulated [12]. In addition, increased cldn-3 expression was also shown to decrease paracellular permeability, which was evaluated by transepithelial electrical resistance and dye flux measurements, but cldn-3 regulation in ALI is still not clearly understood.
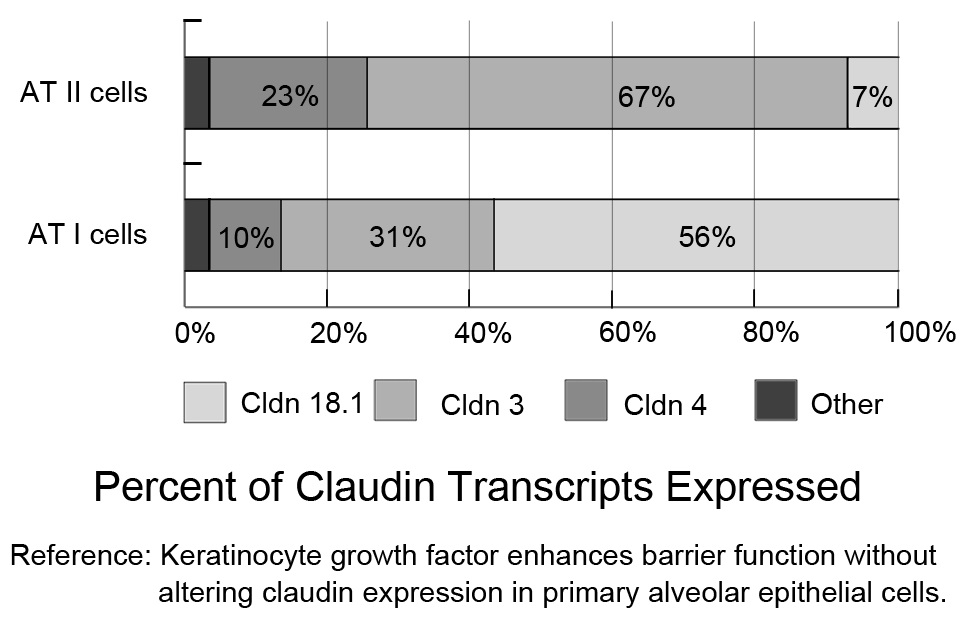 Figure 2: Claudin mRNA expression profiles for Alveolar Type I (AT I) and Alveolar Type II (AT II) cells. Claudins-3, -4, and -18 are the primary claudins expressed in freshly isolated AT I and AT II cells (fluorescence-activated, cell-sorted). Cldn-18.1 is the major transcript in AT I cells, while cldn-3 is predominantly expressed by AT II cells. Both cell types express cldn-4. [9].
Figure 2: Claudin mRNA expression profiles for Alveolar Type I (AT I) and Alveolar Type II (AT II) cells. Claudins-3, -4, and -18 are the primary claudins expressed in freshly isolated AT I and AT II cells (fluorescence-activated, cell-sorted). Cldn-18.1 is the major transcript in AT I cells, while cldn-3 is predominantly expressed by AT II cells. Both cell types express cldn-4. [9].Pulmonary oedema reabsorption depends on active Na+ transport across the alveolar epithelium [13,14]. Primary Na+ transport via the apical surface of AEC, mainly through ENaC and an amiloride-insensitive pathway, and is subsequently pumped out by Na+, K+-ATPase from the basolateral surface into the lung interstitium to form the transepithelial osmotic gradient. This osmotic gradient drives water movement from the alveolar spaces into the alveolar interstitium [13,14].
Sodium reabsorption mediated by ENaC can be inhibited by amiloride, a pyrazine compound. Single-channel studies have shown that at least two forms of amiloride-sensitive ENaC are present in the apical membrane of AT I, AT II (Figure 1) and Foetal Distal Lung Epithelial (FDLE) cells [2,4,6] : the highly selective cation channel (HSC) and Nonselective Cation channel (NSC). These two channels differ in selectivity for Na+ over K+ (HSC with a Na+/K+ selectivity >40; NSC with a Na+/K+ selectivity of ~1.4), unitary conductance (HSC=4-6 pS and NSC=19-24 pS) and other biophysical characteristics [6].
ENaC is composed of three homologous subunits: α, ß, and γ [15], but the total number of subunits for functional channels are uncertain and vary from four (two α-, one ß-, and one γ-subunit) subunits assembled as a tetramer to nine (three α-, three ß-, and three γ-) subunits to create a much larger complex [16]. Edith Hummler and colleagues [17] observed that transgenic neonate mice without the ENaC α-subunit (α-ENaC) die immediately after birth due to impaired lung fluid clearance. Nadia Randrianarison and colleagues [18] have reported that disrupting the ß-ENaC gene locus in mature mice lead to low expression levels of ß-ENaC mRNA and a compensatory increase in α-ENaC and γ-ENaC protein expression, and yet a moderate impairment in baseline AFC was seen. Moreover, sufficient expression of ß-ENaC may be essential to ß2-agonist stimulation of AFC. Pierre M. Barker and colleagues [19] discovered that newborn mice without γ-ENaC had a decreased lung liquid clearance rate. These data indicate that the three ENaC subunits are all indispensable for maximal AFC.
The phenomenon that part of lung liquid resorption could not be inhibited by amiloride was discovered in the lungs of several species including sheep, rabbits, guinea pigs, rats, and humans [16]. As liquid resorption follows the transepithelial osmotic gradient produced by active Na+ transport, there might be an amiloride-insensitive pathway for Na+ transport across the alveolar epithelium. Subsequent studies have indicated that the amiloride-insensitive channel may be a member of CNG cation channel family. CNG cation channels are gated by cAMP or cGMP but not voltage [20,21]. The amiloride-insensitive fraction of Na+ transport-dependent AFC may be partially mediated by CNG cation channels [14]. Ion channels that are directly activated by cyclic nucleotides were first found in the plasma membrane of retinal photoreceptors [21], and now CNG channels have been discovered not only in photoreceptors and olfactory sensory neurons but also in other neurons and non-neuronal tissues, including airway epithelial cells and endothelial cells of the pulmonary artery [22]. CNG cation channels are functionally expressed in adult AT II cells [7], and one of the CNG cation channel isoforms CNGα1 is mainly expressed in rat AT I cells [20]. Activating CNG cation channels with a cGMP analogue, 8Br-cGMP (100 µM), resulted in activated whole-cell cation conductance in isolated rat AT II cells and increased lung liquid clearance in situ. Both effects were still apparent in the presence of amiloride [7,20]. Thus, CNG cation channels are activated by cGMP and may contribute to at least part of the amiloride-insensitive fraction of lung liquid clearance. Although less is known about AFC mediated by CNG cation channels, the potential role of CNG cation channels in lung liquid clearance is worth studying for novel therapeutic strategies for pulmonary oedema.
Na+, K+-ATPase is essential to transepithelial active Na+ transport and exists in the basolateral cell membrane. Na+, K+-ATPase pumps three sodium ions out of the cell and exchanges two potassium ions into the cell [23]. This process consumes 20-30% of Adenosine Tri Phosphate (ATP) at rest to form Na+and K+ gradients across the cell membrane [24]. The Na+ and K+ gradients are essential for maintaining membrane potentials, cell volume and the active transport of other solutes [24]. Four isoforms of the α-subunit and five isoforms of the ß-subunit of Na+, K+-ATPase have been identified [13]. The minimal functional Na+, K+-ATPase is a heterodimer of a single α- and ß-subunit. The α-subunit hydrolyses ATP and provides binding sites for cations. The ß-subunit is necessary for the stability and trafficking of this combination [23]. The α1-, α2-, ß1-isoform of Na+, K+-ATPase (α1-, α2-, ß1-Na+, K+-ATPase) has been discovered in lung epithelial cells [25]. Additionally, α1-Na+, K+-ATPase has been reported to be expressed by AT II cells, α1- and α2-Na+, K+-ATPase can be found on AT I cells [26]. Moreover, α2-Na+, K+-ATPase in AT I cells mediates most of the active Na+ transport and basal lung liquid reabsorption seen in isolated, ouabain-perfused rat lungs [26]. The activity of α1- or α2-Na+, K+-ATPase in these isolated lungs can be selectively inhibited by ouabain at a specific concentration, and the selective inhibitions were used to determine the differential contribution of α1- or α2-Na+, K+-ATPase to AFC ouabain ouabain [26]. In transgenic mice, a 50% decrease in protein expression of either α1- or α2 -Na+, K+-ATPase does not affect basal or stimulated AFC, while a 50% protein loss in both α1 and α2 -Na+, K+-ATPase produces a submaximal cAMP-stimulated AFC without affecting basal AFC [27]. These results indicate that α1- and α2-Na+, K+-ATPase regulate cAMP-stimulated AFC in a coordinated manner.
Regulation of alveolar sodium and fluid transport during ALI
In hypoxic rat AT II cells, hypoxia reduces the transepithelial Na+ current and activity of the amiloride-sensitive Na+ channel, which is related to the hypoxia-induced decrease (quantified by biotinylation) in ENaC subunits, especially the ß- and γ-subunits, in the apical membrane, but hypoxia does not decrease mRNA or protein expression of the ENaC subunits [29]. The agonist. More importantly, terbutaline i ß2-agonist terbutaline reverses the hypoxia-induced down regulation of transepithelial Na+ transport by stimulating Na+ channel activity and increasing the insertion of ENaC subunits into the membrane of hypoxic AECs via cAMP stimulation [29]. In summary, hypoxia inhibits amiloride-sensitive Na+ channel activity by decreasing the apical expression of ENaC subunits, and this inhibition can be reversed by a ß2-agonist. More importantly, terbutaline increases Na+, Cl-; transport in AT II cells during normoxia and hypoxia [30]. However, in isolated rat AT II cells, hypoxia impairs ß2-Adrenergic Receptor (ß2AR) signalling. Although hypoxia decreases terbutaline-stimulated cAMP production and ß2AR density, the potency of terbutaline and affinity between terbutaline and ß2AR are not impacted, and the decrease in cyclic AMP (cAMP) production can be reversed by reoxygenation [31]. ß2AR belongs to a family of G protein-coupled receptors, and the hypoxia-induced downregulation results from increased Gi/o activity [31]. Subsequent research has indicated that prolonged hypoxia impairs ß2AR signalling in alveolar epithelia and whole lungs, but this impairment is no longer aggravated [32]. In addition, alveolar sodium transport under hypoxic conditions can be ameliorated by Dexamethasone (DEX) [33,34].
Cells react to hypoxia through adaptive mechanisms by increasing the expression of genes involved in angiogenesis and glycolytic pathways and maintaining cell ATP homeostasis [35]. As ALI/ARDS is clinically characterised by the abrupt onset of hypoxaemia, about half of patients with ALI/ARDS have died or have given up treatment in 7-10 days [36]. Decreasing the processes that consume ATP, such as Na+, K+-ATPase, is more important for compensation. The exposure of AECs to hypoxia leads to a time-dependent decrease in Na+, K+-ATPase activity via its endocytosis from the plasma membrane into intracellular compartments. Therefore, the amount of α1-Na+, K+-ATPase in the basal cell membrane is decreased, but the total abundance of cellular protein is not [37]. Hypoxia increases the mitochondrial Reactive Oxygen Species (mROS) levels via a capable electron transport chain. Mitochondrial DNA-deficient (ρ0) A549 cells in the absence of the cytochrome oxidase subunit II could not generate hypoxia-induced mROS production and Na+, K+-ATPase endocytosis and decreased Na+, K+-ATPase activity [37]. Meanwhile, antioxidants can inhibit this hypoxia-induced enhancement of mROS and downregulation of Na+, K+-ATPase [37]. Therefore, these results suggest that the endocytosis of Na+, K+-ATPase during hypoxia is mediated by mROS.
In rat AECs, hypoxia-generated mROS leads to the phosphorylation/activation of Adenosine Monophosphate-activated Protein Kinase (AMPK) at the Thr172 residue. The activated AMPK α subunit binds and directly phosphorylates Protein Kinase C (PKCζ) at the Thr410 within the PKCζ activation loop [38]. Moreover, phosphorylation of PKCζ at the Thr410 is required for the hypoxia-induced Na+, K+-ATPase endocytosis. Small interfering RNA knockdown of AMPK α1 but not α2 suggests that PKCζ is specifically activated by AMPK α1 isoform [38]. Activated PKCζ in turn mediates the phosphorylation of Na+, K+-ATPase at Ser18 in the α1-subunit and Na+, K+-ATPase endocytosis but without significant changes in total cell protein abundance [37]. Meanwhile, mROS increases the degradation of Na+, K+-ATPase, which is mediated by the ubiquitin-conjugating system [39]. The connection between the endocytosis and ubiquitination of Na+, K+-ATPase has been described. During hypoxia, ubiquitination of α1-Na+, K+-ATPase has been discovered at the basolateral membrane of AEC and plays an important role in the endocytosis of Na+, K+-ATPase [40]. Both the endocytosis and ubiquitination of α1-Na+, K+-ATPase are prevented when the Ser18 residue in the N-terminus of the α1-subunit (PKCζ phosphorylation motif) is mutated to alanine, suggesting that Ser18 phosphorylation is necessary for these two processes [40]. In addition, mutation of the four lysine residues (K16, K17, K19, K20) adjacent to Ser18 to arginine inhibits the endocytosis and ubiquitination of Na+, K+-ATPase during hypoxia [40]. In conclusion (Figure 3), exposure of AECs to hypoxia leads to AMPK α1 activation by mROS, leading to direct PKCζ phosphorylation at Thr410. Activated PKCζ phosphorylates Na+,K+-ATPase at Ser18 in the α1 subunit, which causes ubiquitination at the sequence KKSKK [four lysine residues (K16, K17, K19, K20) surrounding Ser 18] of α1-Na+, K+-ATPase, leading Na+, K+-ATPase endocytosis. Subsequently, further studies on hypoxia-generated downregulation of Na+, K+-ATPase activity were performed. During hypoxia, mROS also leads to calcium entry through Calcium Release-Activated Calcium (CRAC) channels, which transport stores-operated calcium to replenish endoplasmic reticulum Ca2+ stores in nonexcitable cells [41]. This calcium entry induces Na+, K+-ATPase endocytosis and the phosphorylation/activation of AMPK via Calmodulin-dependent kinase ß (CaMKKß) [41]. Moreover, hypoxia-induced impairment of AFC is prevented when CRAC channel function is inhibited [41]. Thus, during hypoxia, the calcium entry via CRAC channels induces CaMKKß/AMPK activation and Na+, K+-ATPase endocytosis. Additionally, mROS has been shown to possibly play an upstream role in calcium signalling; thus, the calcium flux mediated by CRAC channels may be an intermediary process between mROS-induced AMPK α1 activation and CaMKKß/AMPK activation. Moreover, in H441 human airway epithelial cells, AMPK activation with the AMP mimetic AICAR leads to the inhibition of amiloride-sensitive HSC and NSC, which is associated with a decreased channel open probability [42]. Recent research has shown that hypoxia also activates an AMPK-independent pathway in H441 cells, modification of AMPK avtivity by Lentiviral prevented the effect of hypoxia on Na+, K+-ATPase but not apical amiloride-sensitive Na+ conductance, which different from the regulation of Na+, K+-ATPase and ENaC activity via the above AMPK-dependent pathway [43].
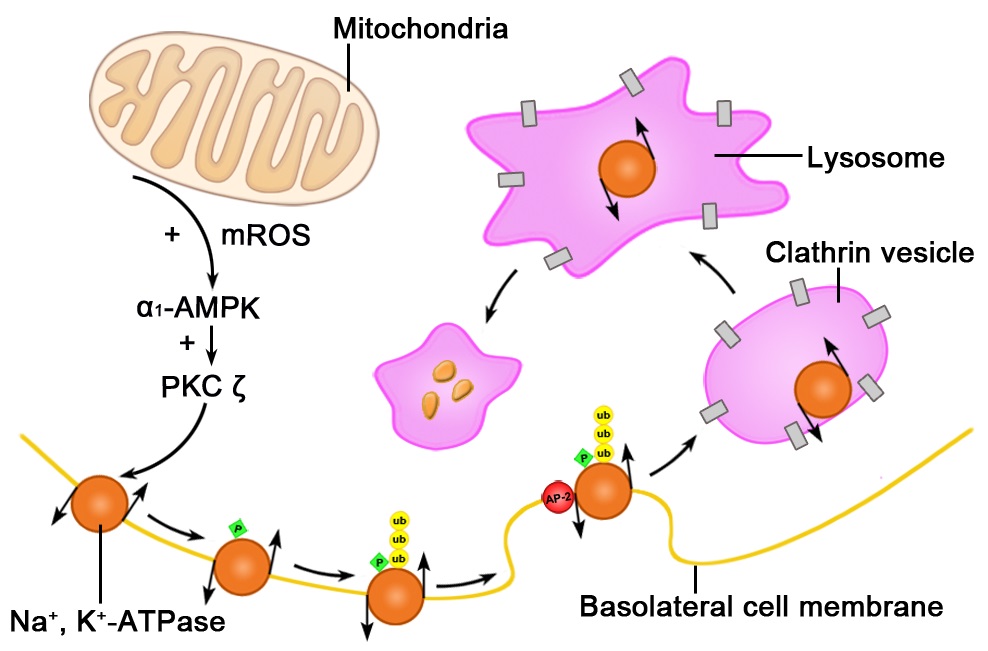 Figure 3: Hypoxia induces the endocytosis and degradation of basolateral membrane sodium-potassium adenosine triphosphatase (Na+, K+-ATPase) in alveolar epithelial cells exposed to hypoxia. Hypoxia-generated mitochondrial reactive oxygen species activate the α subunit of adenosine monophosphate-activated protein kinase (α1-AMPK). Activated α1-AMPK increases protein kinase C activity, which phosphorylates basolateral membrane Na+, K+-ATPase, leading to its ubiquitination. This process leads to the recognition of Na+, K+-ATPase by the Adaptor Protein-2 (AP-2) and subsequent Na+, K+-ATPase endocytosis via clathrin-coated vesicles, which traffic Na+, K+-ATPase to lysosomes for degradation [35,44,45].
Figure 3: Hypoxia induces the endocytosis and degradation of basolateral membrane sodium-potassium adenosine triphosphatase (Na+, K+-ATPase) in alveolar epithelial cells exposed to hypoxia. Hypoxia-generated mitochondrial reactive oxygen species activate the α subunit of adenosine monophosphate-activated protein kinase (α1-AMPK). Activated α1-AMPK increases protein kinase C activity, which phosphorylates basolateral membrane Na+, K+-ATPase, leading to its ubiquitination. This process leads to the recognition of Na+, K+-ATPase by the Adaptor Protein-2 (AP-2) and subsequent Na+, K+-ATPase endocytosis via clathrin-coated vesicles, which traffic Na+, K+-ATPase to lysosomes for degradation [35,44,45].kinase (α1-AMPK). Activated α1-AMPK increases protein kinase C activity, which phosphorylates basolateral membrane Na+, K+-ATPase, leading to its ubiquitination. This process leads to the recognition of Na+, K+-ATPase by the Adaptor Protein-2 (AP-2) and subsequent Na+, K+-ATPase endocytosis via clathrin-coated vesicles, which traffic Na+, K+-ATPase to lysosomes for degradation [35,44,45].
In AECs, hypoxia-induced Na+, K+-ATPase endocytosis requires the binding of Adaptor Protein (AP)-2 to the tyrosine-based motif (Tyr-537) in the α1-subunit of Na+, K+-ATPase, leading to the incorporation of Na+, K+-ATPase into clathrin-coated vesicles [44]. Subsequently, phosphorylated and ubiquitinated Na+, K+-ATPase is degraded in the lysosomes in AECs [45]. Hypoxia-induced degradation of Na+, K+-ATPase is prevented by lysosome and proteasome inhibitors [39].
In addition, mild and severe hypoxia has been shown to lead to a disruption in the cytoskeleton in primary rat AECs, including disorganisation of actin and α-spectrin; moreover, exposure of AECs to mild and severe hypoxia results in the mislocalisation of occludin from the tight junction to the cytoplasm and reduced ZO-1 protein expression [46]. The hypoxia-induced reduction in occludin abundance in the AEC plasma membrane is mediated by PKCζ and protein phosphatase 2A [47]. This downregulation may decrease alveolar Na+ transport through the paracellular pathway.
Regulation of alveolar sodium and fluid transport by RNS and ROS during ALI
Nitric Oxide (NO) modulates lung Na+ transport under both basal conditions and inflammation during ALI [49-53]. Both cGMP-dependent and -independent mechanisms are involved in the regulation of lung Na+transport by NO. NO produced by iNOS under basal conditions is required to regulate amiloride-sensitive Na+ transport via ENaC in the lung epithelium [54,55] because iNOS knockout mice lack amiloride-sensitive Na+ transport across the alveolar and airway epithelia [54]. The specific iNOS inhibitor (1400W) injected intraperitoneally in C57BL/6 mice prevented Na+-dependent amiloride-sensitive AFC [55]. Endogenous NO under basal conditions regulates by the amount of α and γ-ENaC via post-transcriptional, cGMP-independent mechanisms [55]. However, in the H441 human bronchiolar epithelial cell line, using two NO donors, NO was demonstrated to reduce Na+ reabsorption by inhibiting the activity of HSC and Na+, K+-ATPase [53]. In AT II cells, using a NO donor, NO has been suggested to reduce the activity of a predominant apical Na+-permanent cation channel with a conductance of 20.6 ± 1.1 (SE) pS and Na+/K+selectivity of 0.97 ± 0.07 (possible NSC), and this inhibition is mediated by a cGMP-dependent protein kinase [50]. NO reacts with superoxide to form other reactive species, leading to different effects on Na+transport in lung epithelial cells. These effects may partially be responsible for the diverse effects of NO and NO donors on ENaC activity.
in vitro and in vivo results from H441 cells have indicated that PKGII in the NO/cGMP/PKG pathway activates ENaC [56]. Additionally, cGMP has been shown to activate either PKG I or PKGII in cells, and 8-pCPT-cGMP, a PKGII activator, increased amiloride-sensitive short-circuit current (Isc) and whole-cell currents in H441 cells in vitro. These upregulations could be inhibited by Rp-8-pCPT-cGMP, a specific PKGII inhibitor. Consistently, 8-pCPT-cGMP improved amiloride-sensitive AFC in vivo. In addition, Na+/K+-ATPase downregulation by NO or peroxynitrite has also been discovered in the liver, kidneys and lungs [53,57,58].
Peroxynitrite anion (ONOO-), is a reactive oxidant produced from a reaction between NO and superoxide as a response to certain biomolecules, including proteins, lipids and DNA [59]. Physiological concentrations of peroxynitrite reduce AT II cell Na+ transport by impairing apical amiloride-sensitive Na+ channels [59].
In FDLEs, the switch from foetal (3%) to postnatal (21%) O2 concentrations increases ENaC mRNA expression and amiloride-sensitive Na+ transport, and additionally induce Nuclear Factor-?B (NF-?B) [60]. NF-?B and AP-1 response elements have been found in the promoter regions of ENaC subunits [60]. There may be a connection between the upregulation of ENaC gene expression and the O2-induced transcription factor NF-?B. Changes in oxygen concentration also lead to changes in ROS. Because the O2-induced enhancement of ENaC gene expression can be inhibited by the cell-permeable superoxide scavenger tetramethylpiperidine-N-oxyl, the ROS superoxide has been suggested to participate in ENaC regulation [60]. in vivo, using two novel whole animal imaging approaches, ROS were shown to increase ENaC activity and activate lung fluid clearance [61]. Moreover, the balance between superoxide and NO may contribute to alveolar fluid homeostasis. Increasing endogenous superoxide levels using a superoxide dismutase inhibitor (Ethiolat) has been shown to prevent NO inhibition of ENaC activity, which was examined using a single channel patch clamp in AT II cells [62].
Regulation of alveolar sodium and fluid transport by TNF-α during ALI
Therapies for improving alveolar sodium and fluid transport during ALI
Overexpression of the Na+, K+-ATPase subunits via gene transfer shows a potential therapeutic role in improving AFC and epithelial function. Adenovirus-induced gene transfer of the α2-Na+, K+-ATPase gene leads to protein overexpression of α2-Na+, K+-ATPase and increased Na+, K+-ATPase activity in rat AECs and human A549 cells and enhanced α2-Na+, K+-ATPase protein expression in vivo and in ventilation-induced injured lungs, thus improving the basal lung fluid clearance rate [26,68]. ß1-Na+, K+-ATPase over expression via adenovirus-mediated gene transfer has been demonstrated to be efficient for augmenting lung liquid clearance in normal and injured rat lungs [69,70]. By using electroporation, this non viral gene transfer of ß1-Na+, K+-ATPase improves Na+, K+-ATPase activity and AFC [71]. The similar results were observed when using the same experimental technique in lung contusion-induced lung injury. Gene transfer of the a and ß subunits of Na+, K+-ATPase led to improved recovery from ALI [72]. Above all, upregulation of Na+, K+-ATPase via gene transfer of its subunit genes (α2- or ß1-Na+, K+-ATPase) has proven to be useful, even for improving AFC in normal and injured rat lungs.
ß2ARs are distributed throughout the lung [73] and mediate ß-adrenergic stimulation of Na+ and fluid transports [74]. There is no significant difference in the total water content between mice without ß1AR or ß2AR and wild-type controls. ß2AR signalling has been suggested not to be required for basal AFC. However, for physiological adaptive responses, such as hyperoxia, ß2AR signalling is needed for AFC in normal and injured lungs [75]. ß2AR stimulation can increase the activity, expression, and membrane insertion of ENaC and Na+, K+-ATPase and activate CNG cation channels in the alveolar epithelium by increasing intracellular cAMP and cGMP, thus improving AFC [14,31,76]. Moreover, ß2AR-mediated stimulation of Cl-; transport via CFTR also contributes to increased alveolar fluid transport [73]. Both AT I and AT II cells express CFTR and are capable of Cl-;uptake [77]. This interdependency between CFTR and ß2AR is required for ß2AR-stimulated alveolar Na+ and fluid transport [78]. In AECs, ß2-agonists stimulate adenylyl cyclase and subsequently increase intracellular cAMP and activate protein kinase A (PKA). PKA augments the number of HSCs, increases NSC activity, and upregulates Na+, K+-ATPase and CFTR [73,79]. However, FDLEs exposed to the ß2-adrenergic agonist terbutaline have increased Na+transport via a post-transcriptional increase in α1-Na+, K+-ATPase protein expression, without significant effects on ß1-Na+, K+-ATPase and ENaC [25], suggesting that ß2-adrenergic agonists play different roles in ion transport in adult and foetal distal lung epithelium. Furthermore, prolonged treatment with terbutaline impairs ß2AR signalling in the alveolar epithelia and whole lungs [32].
In addition to, other catecholamine, such as dopamine and isoproterenol, have been discovered to potentially upregulate AFC [75,80,81]. Increasing endogenous and clinically administered dopamine improve AFC by upregulating the alveolar epithelial Na+, K+-ATPase [80,81]. Dopamine increases both ENaC and Na+, K+-ATPase activity via different mechanisms [80,82,83]. The former is induced by a cAMP-mediated, alternative signalling pathway [82], and the latter is mediated by Na+, K+-ATPase exocytosis from late endosomes merged into the basolateral membrane of AECs [83].
Although the use of a high-dose Glucocorticoid (GC) for ALI patients remains controversial, DEX enhances the expression of ENaC and Na+, K+-ATPase during both normoxia and hypoxia in rats and primary alveolar AT II cells [33]. GC signalling is mediated by the GC Receptor (GR). Mice with a GR deficiency in all tissues are born dead due to respiratory failure. In lung epithelium-specific GR knockout mice, a reduction in mRNA expression for surfactant proteins, transepithelial Na+ transport, and fluid clearance at birth is seen, which may be involved in decreased viability [84]. Glucocorticoids increase the surface abundance of the ENaC subunits (α-, ß- and γ-ENaC) via a mechanism that may dependent on the activation of Serum and Glucocorticoid-inducible Kinase 1 (SGK1). However, brief but not prolonged exposure of H441 human airway epithelial cells to dexamethasone activates SGK1 and leads to an SGK1-dependent increase in the surface abundance of the ENaC subunits. This upregulation could not explain the persistent activation of ENaC [85]. Even so, SGK1 plays an important role in activating lung ENaC [85,86]. These findings suggest that Na+ transport stimulated by catecholamines and glucocorticoids strengthens AFC and may related to better clinical outcomes for ALI patients.
SUMMARY
Increased pulmonary oedema formation occurs in ALI/ARDS. Patients who failed to remove alveolar oedema fluid rapidly have worse clinical outcomes. AFC depends on vectorial active Na+ transport via apical Na+ channels and basolateral Na+, K+-ATPase and paracellular Na+ transport. Hypoxia in the course of ALI leads to impaired AFC via downregulation of ENaC, Na+, K+-ATPase, and intercellular junctions. The inflammation during ALI contributes to increased RNS, ROS and TNF-α level, which result in different regulation pattern of lung Na+ transport across the alveolar epithelium. However, although these agents play a notable role in ALI, the regulatory mechanisms for alveolar Na+ and fluid transport in ALI are still undefined. Catecholamines and glucocorticoids stimulate Na+ transport and AFC; in particular, ß2-agonists have been shown to play a remarkable role in limiting alveolar oedema in preclinical experimental studies. A single-centre, placebo-controlled small clinical trial reported that sustained treatment with intravenous ß2-agonists reduces extravascular lung fluid in patients with ALI [87]. However, two recent multicentre, placebo-controlled large clinical trials have reported that both intravenous and aerosolised ß2-agonists therapies did not improve clinical outcomes in patients with ALI [88,89]. Routine use of ß2-agonist treatment in mechanically ventilated patients with ALI cannot be recommended. Intravenous ß2-agonists could increase mortality in patients with early ARDS, but the underlying mechanisms of this increase remain unclear. The results of these large controlled clinical trials are more convincing; however, the cause of the different results between preclinical experimental studies or the small controlled clinical trial and the large controlled clinical trials need to be explained.
REFERENCES
- Ware LB, Matthay MA (2001) Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 163: 1376-1383.
- Matalon S, O'Brodovich H (1999) Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu Rev Physiol 61: 627-661.
- Johnson MD, Widdicombe JH, Allen L, Barbry P, Dobbs LG (2002) Alveolar epithelial type I cells contain transport proteins and transport sodium, supporting an active role for type I cells in regulation of lung liquid homeostasis. Proc Natl Acad Sci USA 99: 1966-1971.
- Johnson MD, Bao HF, Helms MN, Chen XJ, Tigue Z, et al. (2006) Functional ion channels in pulmonary alveolar type I cells support a role for type I cells in lung ion transport. Proc Natl Acad Sci USA 103: 4964-4969.
- Borok Z, Liebler JM, Lubman RL, Foster MJ, Zhou B, et al. (2002) Na transport proteins are expressed by rat alveolar epithelial type I cells. Am J Physiol Lung Cell Mol Physiol 282: 599-608.
- Eaton DC, Helms MN, Koval M, Bao HF, Jain L (2009) The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol 71: 403-423.
- Kemp PJ, Kim KJ, Borok Z, Crandall ED (2001) Re-evaluating the Na(+) conductance of adult rat alveolar type II pneumocytes: evidence for the involvement of cGMP-activated cation channels. J Physiol 536: 693-701.
- Overgaard CE, Daugherty BL, Mitchell LA, Koval M ( 2011) Claudins: control of barrier function and regulation in response to oxidant stress. Antioxid Redox Signal 15: 1179-1193.
- LaFemina MJ, Rokkam D, Chandrasena A, Pan J, Bajaj A, et al. (2010) Keratinocyte growth factor enhances barrier function without altering claudin expression in primary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 299: 724-734.
- Mitchell LA, Overgaard CE, Ward C, Margulies SS, Koval M (2011) Differential effects of claudin-3 and claudin-4 on alveolar epithelial barrier function. Am J Physiol Lung Cell Mol Physiol 301: 40-49.
- Wray C, Mao Y, Pan J, Chandrasena A, Piasta F, et al. (2009) Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am J Physiol Lung Cell Mol Physiol 297: 219-227.
- Ohta H, Chiba S, Ebina M, Furuse M, Nukiwa T (2011) Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol 302: 193-205.
- Sznajder JI, Factor P, Ingbar (2002) DH Invited review: lung edema clearance: role of Na(+)-K(+)-ATPase. J Appl Physiol 93: 1860-1866.
- Norlin A, Lu LN, Guggino SE, Matthay MA, Folkesson HG (2001) Contribution of amiloride-insensitive pathways to alveolar fluid clearance in adult rats. J Appl Physiol 90: 1489-1496.
- Canessa CM, Schild L, Buell G, Thorens B, Gautschi I, et al. (1994) Amiloride-sensitive epithelial Na+channel is made of three homologous subunits. Nature 367: 463-467.
- Matthay MA, Folkesson HG, Clerici C (2002) Lung epithelial fluid transport and the resolution of pulmonary edema. Physiol Rev 82: 569-600.
- Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, et al. (1996) Early death due to defective neonatal lung liquid clearance in alphα-ENaC-deficient mice. Nat Genet 12: 325-328.
- Randrianarison N, Clerici C, Ferreira C, Fontayne A, Pradervand S, et al. (2008) Low expression of the betα-ENaC subunit impairs lung fluid clearance in the mouse. Am J Physiol Lung Cell Mol Physiol 294: 409-416.
- Barker PM, Nguyen MS, Gatzy JT, Grubb B, Norman H, et al. (1998) Role of gammaENaC subunit in lung liquid clearance and electrolyte balance in newborn mice. Insights into perinatal adaptation and pseudohypoaldosteronism. J Clin Invest 102: 1634-1640.
- Wilkinson WJ, Benjamin AR, De Proost I, Orogo-Wenn MC, Yamazaki Y, et al. (2011) Alveolar epithelial CNGα1 channels mediate cGMP-stimulated, amiloride-insensitive, lung liquid absorption. Pflugers Arch 462: 267-279.
- Zagotta WN, Siegelbaum SA (1996) Structure and function of cyclic nucleotide-gated channels. Annu Rev Neurosci 19: 235-263.
- Kaupp UB, Seifert R (2002) Cyclic nucleotide-gated ion channels. Physiol Rev 82: 769-824.
- Morth JP, Poulsen H, Toustrup-Jensen MS, Schack VR, Egebjerg J, et al. (2009) The structure of the Na+, K+-ATPase and mapping of isoform differences and disease-related mutations. Philos Trans R Soc Lond B Biol Sci 364: 217-227.
- Jorgensen PL, Hakansson KO, Karlish SJ (2003) Structure and mechanism of Na, K-ATPase: functional sites and their interactions. Annu Rev Physiol 65: 817-849.
- Rahman MS, Gandhi S, Otulakowski G, Duan W, Sarangapani A, et al. (2010) Long-term terbutaline exposure stimulates alphα1-Na+-K+-ATPase expression at posttranscriptional level in rat fetal distal lung epithelial cells. Am J Physiol Lung Cell Mol Physiol 298: 96-104.
- Ridge KM, Olivera WG, Saldias F, Azzam Z, Horowitz S, et al. (2003) Alveolar type 1 cells express the alphα2 Na, K-ATPase, which contributes to lung liquid clearance. Circ Res 92: 453-460.
- Looney MR, Sartori C, Chakraborty S, James PF, Lingrel JB, et al. (2005) Decreased expression of both the alphα1- and alphα2-subunits of the Na-K-ATPase reduces maximal alveolar epithelial fluid clearance. Am J Physiol Lung Cell Mol Physiol 289: 104-110.
- Chen XJ, Seth S, Yue G, Kamat P, Compans RW, et al. (2004) Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol 287: 366-373.
- Planès C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, et al. (2002) Hypoxia and beta 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem 277: 47318-47324.
- Loeh B, Baloglu E, Ke A, Bärtsch P, Mairbäurl H (2010) Betα2-adrenergic stimulation blunts inhibition of epithelial ion transport by hypoxia of rat alveolar epithelial cells. Cell Physiol Biochem 25: 123-134.
- Baloglu E, Ke A, Abu-Taha IH, Bärtsch P, Mairbäurl H (2009) in vitro hypoxia impairs betα2-adrenergic receptor signaling in primary rat alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 296: 500-509.
- Baloglu E, Reingruber T, Bärtsch P, Mairbäurl H (2011) ß2-Adrenergics in hypoxia desensitize receptors but blunt inhibition of reabsorption in rat lungs. Am J Respir Cell Mol Biol 45: 1059-1068.
- Güney S, Schuler A, Ott A, Höschele S, Zügel S, et al. (2007) Dexamethasone prevents transport inhibition by hypoxia in rat lung and alveolar epithelial cells by stimulating activity and expression of Na+-K+-ATPase and epithelial Na+ channels. Am J Physiol Lung Cell Mol Physiol 293: 1332-1338.
- Urner M, Herrmann IK, Booy C, Roth-Z' Graggen B, Maggiorini M, et al. (2012) Effect of hypoxia and dexamethasone on inflammation and ion transporter function in pulmonary cells. Clin Exp Immunol 169: 119-128.
- Zhou G, Dada LA, Sznajder JI (2008) Regulation of alveolar epithelial function by hypoxia. Eur Respir J 31: 1107-1113.
- Wheeler AP, Bernard GR (2007) Acute lung injury and the acute respiratory distress syndrome: a clinical review. Lancet 369: 1553-1564.
- Dada LA, Chandel NS, Ridge KM, Pedemonte C, Bertorello AM, et al. (2003) Hypoxia-induced endocytosis of Na, K-ATPase in alveolar epithelial cells is mediated by mitochondrial reactive oxygen species and PKC-zeta. J Clin Invest 111: 1057-1064.
- Gusarova GA, Dada LA, Kelly AM, Brodie C, Witters LA, et al. (2009) Alphα1-AMP-activated protein kinase regulates hypoxia-induced Na, K-ATPase endocytosis via direct phosphorylation of protein kinase C zeta. Mol Cell Biol 29: 3455-3464.
- Comellas AP, Dada LA, Lecuona E, Pesce LM, Chandel NS, et al. (2006) Hypoxia-mediated degradation of Na, K-ATPase via mitochondrial reactive oxygen species and the ubiquitin-conjugating system. Circ Res 98: 1314-1322.
- Dada LA, Welch LC, Zhou G, Ben-Saadon R, Ciechanover A, et al. (2007) Phosphorylation and ubiquitination are necessary for Na, K-ATPase endocytosis during hypoxia. Cell Signal 19: 1893-1898.
- Gusarova GA, Trejo HE, Dada LA, Briva A, Welch LC, et al. (2011) Hypoxia leads to Na,K-ATPase downregulation via Ca(2+) release-activated Ca(2+) channels and AMPK activation. Mol Cell Biol 31: 3546-3556.
- Albert AP, Woollhead AM, Mace OJ, Baines DL (2008) AICAR decreases the activity of two distinct amiloride-sensitive Na+-permeable channels in H441 human lung epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol 295: 837-848.
- CD Tan, RT Smolenski, MI Harhun, HK Patel, SG Ahmed, et al. (2012) AMP-activated protein Kinase (AMPK)-dependent and -independent pathways regulate hypoxic inhibition of transepithelial Na+transport across human airway epithelial cells. Br J Pharmacol 167: 368-382.
- Zongpei Chen, Rafael T Krmar, Laura Dada, Riad Efendiev, Ingo B Leibiger, et al. (2006) Phosphorylation of adaptor protein-2 µ2 is essential for Na+, K+-ATPase endocytosis in response to either G protein-coupled receptor or reactive oxygen species. Am J Respir Cell Mol Biol 35: 127-132.
- Lecuona E, Sun H, Vohwinkel C, Ciechanover A, Sznajder JI (2009) Ubiquitination participates in the lysosomal degradation of Na, K-ATPase in steady-state conditions. Am J Respir Cell Mol Biol 41: 671-679.
- Bouvry D, Planès C, Malbert-Colas L, Escabasse V, Clerici C (2006) Hypoxia-induced cytoskeleton disruption in alveolar epithelial cells. Am J Respir Cell Mol Biol 35: 519-527.
- Caraballo JC, Yshii C, Butti ML, Westphal W, Borcherding JA, et al. (2011) Hypoxia increases transepithelial electrical conductance and reduces occludin at the plasma membrane in alveolar epithelial cells via PKC- ? and PP2A pathway. Am J Physiol Lung Cell Mol Physiol 300: 569-578.
- Bhatia M, Moochhala S (2004) Role of inflammatory mediators in the pathophysiology of acute respiratory distress syndrome. J Pathol 202: 145-156.
- Hu P, Ischiropoulos H, Beckman JS, Matalon S (1994) Peroxynitrite inhibition of oxygen consumption and sodium transport in alveolar type II cells. Am J Physiol 266: 628-634.
- Jain L, Chen XJ, Brown LA, Eaton DC (1998) Nitric oxide inhibits lung sodium transport through a cGMP-mediated inhibition of epithelial cation channels. Am J Physiol 274: 475-484.
- Hickman-Davis JM, McNicholas-Bevensee C, Davis IC, Ma HP, Davis GC, et al. (2006) Reactive species mediate inhibition of alveolar type II sodium transport during mycoplasma infection. Am J Respir Crit Care Med 173: 334-344.
- Nielsen VG, Baird MS, Chen L, Matalon S (2000) DETANONOate, a nitric oxide donor, decreases amiloride-sensitive alveolar fluid clearance in rabbits. Am J Respir Crit Care Med 161: 1154-1160.
- Althaus M, Pichl A, Clauss WG, Seeger W, Fronius M, et al. (2010) Nitric oxide inhibits highly selective sodium channels and the Na+/K+-ATPase in H441 cells. Am J Respir Cell Mol Biol 44: 53-65.
- Hardiman KM, Lindsey JR, Matalon S (2001) Lack of amiloride-sensitive transport across alveolar and respiratory epithelium of iNOS(-/-) mice in vivo. Am J Physiol Lung Cell Mol Physiol 281: 722-731.
- Hardiman KM, McNicholas-Bevensee CM, Fortenberry J, Myles CT, Malik B, et al. (2004) Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am J Respir Cell Mol Biol 30: 720-728.
- Nie HG, Chen L, Han DY, Li J, Song WF, et al. (2009) Regulation of epithelial sodium channels by cGMP/PKGII. J Physiol 587: 2663-2676.
- Muriel P, Sandoval G (2000) Nitric oxide and peroxynitrite anion modulate liver plasma membrane fluidity and Na(+)/K(+)-ATPase activity. Nitric Oxide 4: 333-342.
- Zhang C, Imam SZ, Ali SF, Mayeux PR (2002) Peroxynitrite and the regulation of Na(+), K(+)-ATPase activity by angiotensin II in the rat proximal tubule. Nitric Oxide 7: 30-35.
- Szabó C (2003) Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett 140-141: 105-112.
- Rafii B, Tanswell AK, Otulakowski G, Pitkänen O, Belcastro-Taylor R, et al. (1998) O2-induced ENaC expression is associated with NF-kappaB activation and blocked by superoxide scavenger. Am J Physiol 275: 764-770.
- Goodson P, Kumar A, Jain L, Kundu K, Murthy N, et al. (2012) Nadph oxidase regulates alveolar epithelial sodium channel activity and lung fluid balance in vivo via O-2 signaling. Am J Physiol Lung Cell Mol Physiol 302: 410-419.
- Helms M, Jain L, Self JL, Eaton DC (2008) Redox regulation of epithelial sodium channels examined in alveolar type 1 and 2 cells patch-clamped in lung slice tissue. J Biol Chem 283: 22875-22883.
- Yamagata T, Yamagata Y, Nishimoto T, Hirano T, Nakanishi M, et al. (2009) The regulation of amiloride-sensitive epithelial sodium channels by tumor necrosis factor-αlpha in injured lungs and alveolar type II cells. Respir Physiol Neurobiol 166: 16-23.
- Dagenais A, Fréchette R, Clermont ME, Massé C, Privé A, et al. (2006) Dexamethasone inhibits the action of TNF on ENaC expression and activity. Am J Physiol Lung Cell Mol Physiol 291: 1220-1231.
- Vadász I, Schermuly RT, Ghofrani HA, Rummel S, Wehner S, et al. (2008) The lectin-like domain of tumor necrosis factor-αlpha improves alveolar fluid balance in injured isolated rabbit lungs. Crit Care Med 36: 1543-1550.
- Fukuda N, Jayr C, Lazrak A, Wang Y, Lucas R, et al. (2001) Mechanisms of TNF-αlpha stimulation of amiloride-sensitive sodium transport across alveolar epithelium. Am J Physiol Lung Cell Mol Physiol 280: 1258-1265.
- Elia N, Tapponnier M, Matthay MA, Hamacher J, Pache JC, et al. (2003) Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-αlpha. Am J Respir Crit Care Med 168: 1043-1050.
- Adir Y, Welch LC, Dumasius V, Factor P, Sznajder JI, et al. (2008) Overexpression of the Na-K-ATPase alphα2-subunit improves lung liquid clearance during ventilation-induced lung injury. Am J Physiol Lung Cell Mol Physiol 294: 1233-1237.
- Factor P, Dumasius V, Saldias F, Brown LA, Sznajder JI (2000) Adenovirus-mediated transfer of an Na+/K+-ATPase betα1 subunit gene improves alveolar fluid clearance and survival in hyperoxic rats. Hum Gene Ther 11: 2231-2242.
- Factor P, Saldias F, Ridge K, Dumasius V, Zabner J, et al. (1998) Augmentation of lung liquid clearance via adenovirus-mediated transfer of a Na, K-ATPase betα1 subunit gene. J Clin Invest 102: 1421-1430.
- Machado-Aranda D, Adir Y, Young JL, Briva A, Budinger GR, et al. (2005) Gene transfer of the Na+, K+-ATPase betα1 subunit using electroporation increases lung liquid clearance. Am J Respir Crit Care Med 171: 204-211.
- Machado-Aranda DA, Suresh MV, Yu B, Raghavendran K (2012) Electroporation-mediated in vivogene delivery of the Na+/K+-ATPase pump reduced lung injury in a mouse model of lung contusion. J Trauma Acute Care Surg 72: 32-39.
- Mutlu GM, Factor P (2008) Alveolar epithelial betα2-adrenergic receptors. Am J Respir Cell Mol Biol 38: 127-134.
- Barnard ML, Olivera WG, Rutschman DM, Bertorello AM, Katz AI, et al. (1997) Dopamine stimulates sodium transport and liquid clearance in rat lung epithelium. Am J Respir Crit Care Med 156: 709-714.
- Mutlu GM, Dumasius V, Burhop J, McShane PJ, Meng FJ, et al. (2004) Upregulation of alveolar epithelial active Na+ transport is dependent on betα2-adrenergic receptor signaling. Circ Res 94: 1091-1100.
- Wu X, Li S, Wang X, Song Y, Chen Z, et al. (2011) Therapeutic role of terbutaline in a rat whole-lung lavage model. Exp Lung Res 37: 542-548.
- Johnson M, Allen L, Dobbs L (2009) Characteristics of Cl- uptake in rat alveolar type I cells. Am J Physiol Lung Cell Mol Physiol 297: 816-827.
- Mutlu GM, Adir Y, Jameel M, Akhmedov AT, Welch L, et al. (2005) Interdependency of beta-adrenergic receptors and CFTR in regulation of alveolar active Na+ transport. Circ Res 96: 999-1005.
- Chen XJ, Eaton DC, Jain L (2002) Beta-adrenergic regulation of amiloride-sensitive lung sodium channels. Am J Physiol Lung Cell Mol Physiol 282: 609-620.
- Saldías FJ, Comellas AP, Pesce L, Lecuona E, Sznajder JI (2002) Dopamine increases lung liquid clearance during mechanical ventilation. Am J Physiol Lung Cell Mol Physiol 283: 136-143.
- Adir Y, Azzam ZS, Lecuona E, Leal S, Pesce L, et al. (2004) Augmentation of endogenous dopamine production increases lung liquid clearance. Am J Respir Crit Care Med 169: 757-763.
- Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L (2006) Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol 290: 710-722.
- Ridge KM, Dada L, Lecuona E, Bertorello AM, Katz AI, et al. (2002) Dopamine-induced exocytosis of Na, K-ATPase is dependent on activation of protein kinase C-epsilon and -delta. Mol Biol Cell 13: 1381-1389.
- Manwani N, Gagnon S, Post M, Joza S, Muglia L, et al. (2010) Reduced viability of mice with lung epithelial-specific knockout of glucocorticoid receptor. Am J Respir Cell Mol Biol 43: 599-606.
- Watt GB, Ismail NA, Caballero AG, Land SC, Wilson SM (2012) Epithelial Na+ channel activity in human airway epithelial cells: the role of serum and glucocorticoid-inducible kinase 1. Br J Pharmacol 166: 1272-1289.
- Zhu T, Zhang W, Wang DX (2012) Insulin up-regulates epithelial sodium channel in LPS-induced acute lung injury model in rats by SGK1 activation. Injury 43: 1277-1283.
- Perkins GD, McAuley DF, Thickett DR, Gao F (2006) The Beta-Agonist Lung Injury Trial (BALTI): a randomized placebo-controlled clinical trial. Am J Respir Crit Care Med 173: 281-287.
- Matthay MA, Brower RG, Carson S, Douglas IS, Eisner M, et al. (2011) Randomized, placebo-controlled clinical trial of an aerosolized ß2-agonist for treatment of acute lung injury. Am J Respir Crit Care Med 184: 561-568.
- Gao Smith F, Perkins GD, Gates S, Young D, McAuley DF, et al. (2012) Effect of intravenous ß-2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI-2): a multicentre, randomised controlled trial. Lancet 379: 229-235.
Citation: Gao G, Dong C, Yuan M, Liu H, Li B (2014) Sodium Transport and Alveolar Fluid Clearance in Acute Lung Injury. J Cytol Tissue Biol 1: 003.
Copyright: © 2014 Guangyuan Gao, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.