Supplemental Fetal Calf Serum in T cell Culture Media Expands Bovine Serum Albumin-Specific Human CD4+ T Cells
*Corresponding Author(s):
Weisan ChenDepartment Of Biochemistry And Genetics, La Trobe Institute For Molecular Science, La Trobe University, Bundoora, VIC, Australia
Email:weisan.chen@latrobe.edu.au
Abstract
Ex vivo or in vitro expansion of antigen-specific T cells are usually required for clinical trial monitoring of T cell responses after vaccination or immunotherapy. To be able to proliferate, antigen-specific T cells need to recognize antigens presented on the surface of antigen presenting cells (APC) for activation. Fetal calf serum (FCS) is used in many laboratories worldwide as a supplement to T cell culture media. We found that Epstein-Barr virus (EBV)-transformed autologous B cells (BCLs) cultured in FCS-containing, not in human serum-containing or serum-free media, activated CD4+ T cells from human peripheral blood mononuclear cells (PBMCs). We successfully identified bovine serum albumin (BSA), the most abundant protein in FCS, as the antigen that stimulated its specific CD4+ T cells restricted to HLA-DRB1*01:01 and HLA-DPB1*03:01. Furthermore, of the 101 synthetic overlapping BSA peptides screened, two HLA-DRB1*01:01-restricted epitopes were identified. Thus, caution should be exercised when FCS-containing culture media is used for T cell clinical trial monitoring.
Introduction
Antigen-specific CD4+ T cells have the potential to sustain effective immune responses to pathogens and cancers. In the response to cancers, tumor-specific CD4+ T cells secrete interleukin 2 (IL-2) that maintains the survival and function of tumor-specific CD8+ T cells or upregulating CD40 ligand that matures professional antigen presenting cells (APCs) to activate CD8+ T cells [1, 2]. In addition, tumor-infiltrating CD4+ T cells also express multiple effector/cytolytic molecules [3], suggesting their direct effector function. Indeed, adoptive transfer of tumor antigen-specific CD4+ T cells successfully eradicated melanoma [4]. Moreover, adoptive transfer of antigen-specific CD4+ regulatory T cells (Tregs), which constantly express transcription factor Foxp3, is considered a promising immunotherapy to treat autoimmune diseases [5, 6].
Long-term culture is usually required to expand considerable numbers of antigen-specific T cells for clinical trial monitoring of T cell responses after vaccination or immunotherapy. However, development of optimal antigen-specific T cell culture systems has been a major challenge to the clinical monitoring of T cell responses. Antigen-specific T cells are often expanded by specific antigen-pulsed APCs. Interestingly, before being pulsed with the antigens of interest, APCs are often exposed to various irrelevant antigens, including antigens derived from fetal calf serum (FCS), which is supplemented to cell culture or cell isolation media [7-12]. Thus, it is highly likely that APCs could capture and present such antigens to T cells.
In this study, we show that human CD4+ T cells from healthy donor PBMCs (peripheral blood mononuclear cells) were activated by Epstein-Barr virus (EBV)-transformed autologous B cells (BCLs) in RPMI 1640 media containing 10% FCS (RF10). We performed intracellular cytokine production assay using APCs maintained in FCS-containing or -free media to define the antigen’s FCS origin. We eventually identified serum protein bovine serum albumin (BSA) as the antigen to activate its specific CD4+ T cells. Using a combination of anti-HLA (human leukocyte antigen) antibodies and a BCL panel that matched a single donor’s HLA molecules, we were able to define these HLA-DRB1*01:01- and HLA-DPB1*03:01-restricted CD4+ T cell responses. Finally, in vitro stimulating these CD4+ T cells with synthetic overlapping BSA peptides, which together covered the full BSA amino acid (aa) sequences, allowed us to identify two HLA-DRB1*01:01-restricted epitopes.
Our data demonstrate for the first time that APCs cultured in media containing FCS can process BSA and present its antigenic peptides to human CD4+ T cells, which can be a matter of concern for clinical trial monitoring of T cell immunotherapy.
Methods and Materials
Ethics statement
Healthy donor blood buffy packs were obtained from Australian Red Cross Blood Service (Melbourne, Australia) under the agreement 12-07VIC-17. The study was approved by the La Trobe University Faculty Human Ethics Committee under project number FHEC12/NR81.
Antibodies and synthetic peptides
Fluorescence-conjugated mouse anti-human CD3 (V450), CD4 (APC), CD107a (FITC), IFN-γ (FITC or PerCP-Cy5.5), IL-2 (PE), TNF-α (PE-Cy7) mAbs were purchased from BD Biosciences (USA). Anti-rVV RNA-binding protein antibody TW2.3 [13] was a kind gift from Drs Jonathan Yewdell and Jack Bennink (National Institutes of Health, Bethesda, MD). BSA overlapping 18-mers with 6-aa shifts were synthesized as cleaved pin peptides (peptide sequences are shown in supplementary table 1) by Mimotopes (Clayton, Victoria, Australia). All peptides were dissolved in dimethyl sulfoxide (DMSO), aliquoted and stored at -20°C until use.
PBMC isolation
Human PBMCs were isolated via Ficoll-Paque separation and stored in liquid nitrogen until use. All blood donors were HLA typed by Victorian Transplantation and Immunogenetics Service (Melbourne, Australia).
Cell culture
BCLs were generated via standard EBV transformation and cultured in RF10 [RPMI 1640 media (Gibco, USA) supplemented with 10% FCS (Hyclone, USA), 50 μM 2-mercaptoethanol (Gibco, USA) and 100 U/mL Penicillin-Streptomycin (Gibco, USA)]. CD4+ T cells isolated from human PBMCs via flow cytometry cell sorting were cultured in RF10 supplemented with 1× sodium pyruvate (Gibco, USA), 1× non-essential amino acid (Gibco, USA) and 20 U/mL recombinant human interleukin 2 (rhIL-2) (PeproTech, USA). For T cell stimulation and expansion, resting CD4+ T cells were stimulated with irradiated BCLs at a ratio of 10:1, cell culture media were replaced every second day or when the culture media turned yellow.
Preparation of rVV-EBV-infected BCL lysates
BCLs were seeded in 24-well cell culture plate and infected with recombinant vaccinia virus (rVV) expressing individual EBV latent antigen (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, EBNA-LP, LPM1 and LMP2) [14] at a multiplicity of infection (MOI) of 20 for 1 h at 37°C in 200 μL phosphate-buffered saline (PBS) containing 0.1% BSA (Sigma-Aldrich, USA). This was followed by an addition of 1.8 mL RF-10 and overnight incubation at 37°C with 5% CO2. The infected BCLs were pelleted and lysed with 8 M urea [15]. The lysates were then aliquoted and stored at -20°C until use.
Cell staining and flow cytometry
For intracellular staining of TNF-α, IL-2 and IFN-γ in T cells, T cells were cultured alone or cocultured with APCs with or without peptides (10 µg/mL) for 5 h in the presence of 10 µg/mL brefeldin A (BFA) (Sigma-Aldrich, USA). Cells were then washed once with phosphate-buffered saline (PBS), stained for surface markers CD3 and CD4 for 30 min at 4°C, fixed with 1% paraformaldehyde (PFA) for 20 min at room temperature (RT), permeabilized with 2% saponin and stained with mAbs to TNF-α, IL-2 and IFN-γ. For CD107a staining, T cells were antigen-stimulated at 37°C for 5h in the presence of monensin and anti-CD107a antibody before being harvested and stained with anti-CD3 and anti-CD4 mAbs. For T-cell receptor (TCR) V beta (Vβ) chain staining, resting T cells were stained with a panel of Vβ antibodies at 4°C for 30 min using a TCR Vβ repertoire kit (Beckman Coulter Life Sciences, USA). After staining, samples were acquired on a FACS Canto II flow cytometer (Becton Dickinson, USA). The data were analysed with FlowJo software (FlowJo VX, Ashland, OR, USA).
Cell sorting
Resting CD4+ T cells were stained with a panel of Vβ antibodies at 4°C for 30 min, followed by flow cytometry cell sorting of Vβ positive cells using a BD FACSAria III cell sorter. Purity of sorted cells was > 95%.
HLA restriction assay
To determine the presenting HLA class II (HLA-II) alleles, autologous BCLs were blocked with pan-HLA-II specific antibodies for 30 min at 4°C before coculture with T cells. Alternatively, HLA-II restriction was determined via antigen presentation assays using APCs with partial-matched or mismatched HLA-II alleles.
CFSE labelling of responder cells
Responder Vβ1+CD4+ T cells were labelled with carboxyfluorescein succinimidyl ester (CFSE) [20] (Thermo Fisher, USA) before co-culture with PBMC to differentiate from CD4+ T cells from the added PBMC. In brief, CFSE was diluted into 0.08 μM staining solution with PBS. Vβ1+CD4+ T cells were then resuspended in 1 mL of the solution and incubated at 37 °C for 10 min. Labelling was quenched by adding into an equal volume of heat-inactivated FCS. The CFSE-labelled Vβ1+CD4+ T cells were washed 3 times with PBS and resuspended in RPMI-1640 media until use.
Statistical analysis
Statistical analysis was performed using Prism 8 (GraphPad Software). Statistically significant differences between groups were determined by the Student’s t-test: ***P < 0.001, ****P < 0.0001. The error bars indicate the standard error of the mean.
Results
BCLs cultured in media containing FCS activate and expand a population of CD4+ T cells from healthy donor’s PBMCs
To overcome the difficulty in obtaining autologous APCs, EBV-transformed BCLs are often established. Such BCLs can be used to propagate autologous T cells specific to antigens of interest. In this study, BCLs from a healthy donor (2013donor5) were generated, and cocultured with autologous PBMCs in RF10 as a control group. Strikingly, without additional antigens, the BCLs were able to expand autologous CD4+ T cells, as evidenced by the rapid CD3 down-regulation and CD107a up-regulation on stimulated CD4+ T cells 5 h after restimulation (Figures 1A-1B). Moreover, the activated CD4+ T cells formed clusters in the culture (Figures 1C), a typical morphological change of antigen-specific T cells after activation. We further looked into profiles of TNF-α, IL-2 and IFN-γ production after stimulation of these CD4+ T cells with autologous BCLs. It was clear that nearly all T cells that produced IL-2 were positive for TNF-α (Figures 1D-1E). When this analysis was extended to IFN-γ, the data showed that almost all IFN-γ-producing cells also produced TNF-α and IL-2 (Figures 1D -1E). This indicates a distinct TNF-α > IL-2 > IFN-γ cytokine hierarchy, which contrasts with the classic IFN-γ > TNF-α > IL-2 cytokine hierarchy observed in the murine system previously [16, 17]. Taken together, these data suggest that autologous BCLs cultured in FCS-containing media trigger some CD4+ T cells from PBMCs to proliferate and produce TNF-α, IL-2 and IFN-γ in a distinct cytokine hierarchy.
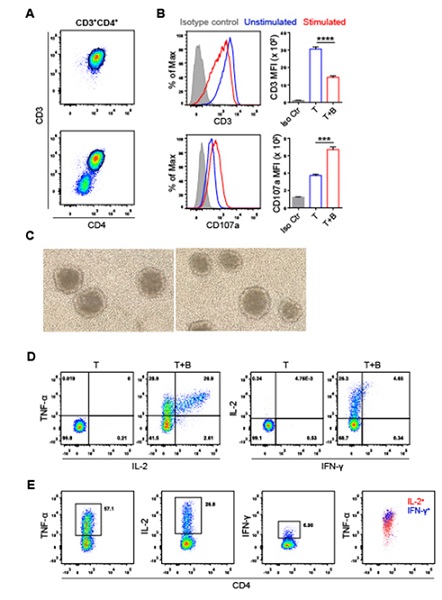 Figure 1: Autologous BCLs cultured in media containing FCS activate human CD4+ T cells. (A) Flow cytometry gating for CD4+ T cells with (bottom) or without (top) stimulation by FCS-exposed autologous BCLs. CD4 T cells were gated based on their cell surface expression of CD3 and CD4. (B) Histograms and bar graphs show the levels of CD3 or CD107a expression in unstimulated and stimulated CD4+T cells, with antibody isotype control shown. Statistical significance was determined with the Student’s t-test: ***P <0.001, ****P < 0.0001. The error bars indicate the standard error of the mean. (C) CD4+ T cells form clusters in the culture after stimulation by FCS-exposed autologous BCLs. (D) Representative FACS plots of CD4+ T cells producing TNF-α, IL-2 and IFN-γ after stimulation. (E) The stimulated CD4+ T cells produced TNF-α, IL-2 and IFN-γ in a cytokine hierarchy of TNF-α > IL-2 > IFN-γ.
Figure 1: Autologous BCLs cultured in media containing FCS activate human CD4+ T cells. (A) Flow cytometry gating for CD4+ T cells with (bottom) or without (top) stimulation by FCS-exposed autologous BCLs. CD4 T cells were gated based on their cell surface expression of CD3 and CD4. (B) Histograms and bar graphs show the levels of CD3 or CD107a expression in unstimulated and stimulated CD4+T cells, with antibody isotype control shown. Statistical significance was determined with the Student’s t-test: ***P <0.001, ****P < 0.0001. The error bars indicate the standard error of the mean. (C) CD4+ T cells form clusters in the culture after stimulation by FCS-exposed autologous BCLs. (D) Representative FACS plots of CD4+ T cells producing TNF-α, IL-2 and IFN-γ after stimulation. (E) The stimulated CD4+ T cells produced TNF-α, IL-2 and IFN-γ in a cytokine hierarchy of TNF-α > IL-2 > IFN-γ.
CD4+ T cells respond specifically to BSA in FCS
We next tried to identify the potential antigens recognized by these CD4+ T cells. FCS was the first to be tested since it was the major soluble protein source the BCLs were exposed to in the culture media. To understand the kinetics of FCS-derived antigen presentation, BCLs cultured in FCS-containing media were shifted into FCS-free media (AIM-V) (Gibico, USA) daily for 4 days and on day 7 to monitor their tailing antigen presentation to the responding CD4+ T cells. As expected, the antigen presentation diminished gradually, reaching baseline after 4 days (Supplementary figure 1A). Thus, BCLs were cultured in AIM-V media for 4 days and then transferred into AIM-V media containing a series of two-fold diluted FCS. After overnight incubation, BCLs were cocultured with the CD4+ T cells, followed by the assessment of the production of TNF-α and IL-2, the most abundant cytokines produced by these CD4+ T cells. Clearly, BCLs stimulated CD4+ T cells to produce TNF-α and IL-2 in an FCS-dose-dependent manner (Figures 2A and 2C), indicating CD4+ T cell recognition of FCS-derived antigens. We further tested bovine serum albumin (BSA), the most abundant serum protein. Interestingly, some of the CD4+ T cells were stimulated by BCLs cultured overnight in BSA-containing AIM-V media in a BSA-dose-dependent manner (Figures 2B and 2D). Of note, BCLs cultured in AIM-V media containing 0.625-1.25% BSA were capable of stimulating a CD4+ T cell response to an equal magnitude as BCLs cultured in AIM-V media containing 10% FCS did, indicating that BSA was the protein captured, processed and presented by BCLs to these CD4+ T cells. Considering amino acid sequences of BSA and human serum albumin (HSA) have 76% similarity [18], we also tested CD4+ T cell response to human serum (HS). Firstly, BCLs were cultured in AIM-V media for 4 or 7 days, followed by overnight culture in AIM-V media supplemented with 10% HS. The CD4+ T cells were then cocultured with the BCLs and intracellularly stained for TNF-α. As expected, these BSA-specific CD4+ T cells failed to respond to HSA (Supplementary figure 1B).
Since BCLs were generated via standard EBV transformation, it remained possible that EBV antigens from latent proteins could be presented to specific CD4+ T cells as endogenous EBV antigen has been reported to be presented by class II molecules [19]. To test this hypothesis, we infected BCLs with a panel of 8 rVVs encoding individual EBV latent proteins, including EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, EBNA-LP, LPM1 and LMP2 [14, 20] (Supplementary figure 2A). The infected BCLs were then harvested and lysed to serve as soluble antigen sources. BCLs cultured in AIM-V media for 4 days were then incubated with such EBV antigen overnight before being cocultured with the CD4+ T cells. Interestingly, BCLs presenting individual EBV latent protein-derived antigens failed to activate any of the CD4+ T cells (Supplementary figure 2B), indicating these CD4+ T cells were not EBV specific. This was further confirmed by an EBV-free melanoma cell line MEL-14 bearing HLA-DR*01:01, which was capable of stimulating these CD4+ T cells to produce TNF-α only when antigen was provided (Supplementary figures 3A-3B). Collectively, these results indicate that the autologous BCL-expanded CD4+ T cells are specific for BSA.
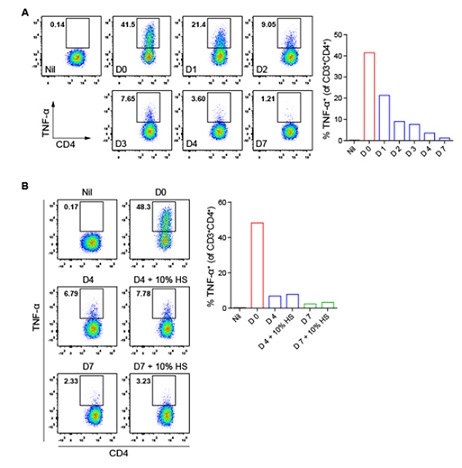 Supplementary Figure 1: Autologous BCLs cultured in FCS-free or HS-containing medium fail to stimulate FCS-specific CD4+ T cells. (A) TNF-α production in CD4+ T cells after stimulation by FCS-free medium-grown autologous BCLs. The BCLs were cultured in FCS-containing medium (Day 0) or in FCS-free medium, AIM-V, for up to 7 days before being used to stimulate FCS-specific CD4+ T cells. (B) CD4+ T cell production of TNF-α in response to FCS-or HS-exposed autologous BCLs. Autologous BCLs were cultured in FCS-containing medium (Day 0), or in FCS-containing media after being cultured in AIM-V media for 4 or 7 days (“day 4 + 10% HS” or “day 7 + 10% HS”)before CD4+ T cell stimulation. Nil shows TNF-α production in unstimulated CD4+ T cells that were cultured alone (A and B).
Supplementary Figure 1: Autologous BCLs cultured in FCS-free or HS-containing medium fail to stimulate FCS-specific CD4+ T cells. (A) TNF-α production in CD4+ T cells after stimulation by FCS-free medium-grown autologous BCLs. The BCLs were cultured in FCS-containing medium (Day 0) or in FCS-free medium, AIM-V, for up to 7 days before being used to stimulate FCS-specific CD4+ T cells. (B) CD4+ T cell production of TNF-α in response to FCS-or HS-exposed autologous BCLs. Autologous BCLs were cultured in FCS-containing medium (Day 0), or in FCS-containing media after being cultured in AIM-V media for 4 or 7 days (“day 4 + 10% HS” or “day 7 + 10% HS”)before CD4+ T cell stimulation. Nil shows TNF-α production in unstimulated CD4+ T cells that were cultured alone (A and B).
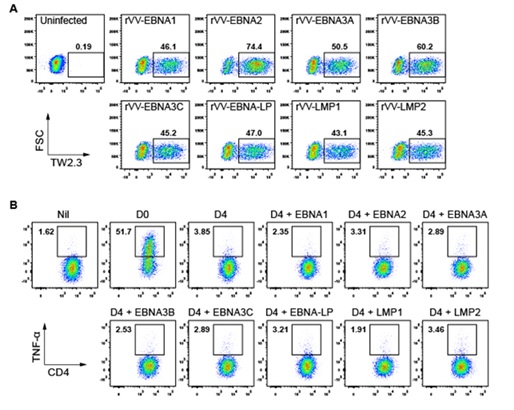 Supplementary Figure 2: CD4+ T cells do not respond to EBV antigens. (A) rVV-EBV infection of autologous BCLs. Autologous BCLs were infected by 8 rVV encoding individual EBV latency proteins, including EBNA1, EBNA2,EBNA3A, EBNA3B, EBNA3C, EBNA-LP, LPM1 and LMP2. Representative FACS plots show the percentages of infected cells recognized by anti-rVV RNA-binding protein antibody TW2.3. (B) TNF-α production in unstimulated CD4+ T cells (Nil) and in the CD4+ T cells stimulated by FCS-exposed autologous BCLs or autologous BCLs pulsed with rVV-EBV-infected BCL lysate.
Supplementary Figure 2: CD4+ T cells do not respond to EBV antigens. (A) rVV-EBV infection of autologous BCLs. Autologous BCLs were infected by 8 rVV encoding individual EBV latency proteins, including EBNA1, EBNA2,EBNA3A, EBNA3B, EBNA3C, EBNA-LP, LPM1 and LMP2. Representative FACS plots show the percentages of infected cells recognized by anti-rVV RNA-binding protein antibody TW2.3. (B) TNF-α production in unstimulated CD4+ T cells (Nil) and in the CD4+ T cells stimulated by FCS-exposed autologous BCLs or autologous BCLs pulsed with rVV-EBV-infected BCL lysate.
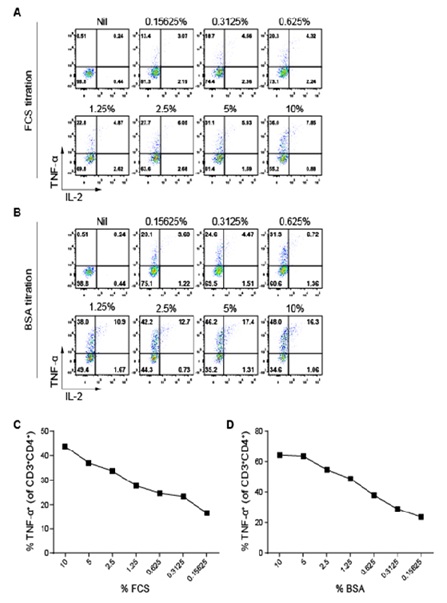 Figure 2: CD4+ T cells produce TNF-α and IL-2 in response to FCS and BSA in a dose-dependent manner. AIM-V medium-grown autologous BCLs were cultured overnight in media containing various FCS or BSA concentrations, and stimulated CD4+ T cells were enumerated via ICS. (A and B) Representative FACS plots of TNF-α and IL-2 expressions in the unstimulated (Nil) and stimulated CD3+CD4+ T cells. (C and D) Percentages of TNF-α+ cells in the stimulated CD3+CD4+ T cells.
Figure 2: CD4+ T cells produce TNF-α and IL-2 in response to FCS and BSA in a dose-dependent manner. AIM-V medium-grown autologous BCLs were cultured overnight in media containing various FCS or BSA concentrations, and stimulated CD4+ T cells were enumerated via ICS. (A and B) Representative FACS plots of TNF-α and IL-2 expressions in the unstimulated (Nil) and stimulated CD3+CD4+ T cells. (C and D) Percentages of TNF-α+ cells in the stimulated CD3+CD4+ T cells.
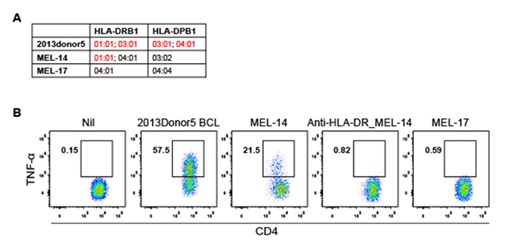 Supplementary figure 3. CD4+ T cells can be stimulated by FCS-exposed HLA-DRB1*01:01-expressing allogenic melanoma cell line. (A) Expression of HLA-DR and HLA-DP molecules on autologous 2013donor5 BCLs or two allogenic melanoma cell lines (MEL-14 and MEL-17). (B) FACS plots of TNF-α production in in unstimulated CD4+ T cells (Nil) and in the CD4+ T cells stimulated by FCS-exposed autologous BCLs, MEL-14 or MEL-17, or anti-HLA-DR antibody-treated MEL-14.
Supplementary figure 3. CD4+ T cells can be stimulated by FCS-exposed HLA-DRB1*01:01-expressing allogenic melanoma cell line. (A) Expression of HLA-DR and HLA-DP molecules on autologous 2013donor5 BCLs or two allogenic melanoma cell lines (MEL-14 and MEL-17). (B) FACS plots of TNF-α production in in unstimulated CD4+ T cells (Nil) and in the CD4+ T cells stimulated by FCS-exposed autologous BCLs, MEL-14 or MEL-17, or anti-HLA-DR antibody-treated MEL-14.
The responses of BSA-specific CD4+ T cells are HLA-DRB1*01:01- and HLA-DPB1*03:01-restricted
First, we performed an HLA blocking assay using antibodies against pan-HLA class II molecules HLA-DR, -DP and -DQ to identify the molecules by which the response of the CD4+ T cells is restricted. The results showed that anti-HLA-DQ did not inhibit CD4+ T cell response (Figure 3A). However, the response was clearly blocked by Anti-HLA-DR and -DP antibodies (Figure 3A), suggesting that the bulk CD4+ T cell responses were HLA-DR and -DP restricted. We next identified the restricting HLA-DR and -DP alleles. As the autologous BCLs expressed HLA-DRB1*01:01, 03:01 and HLA-DPB1*03:01, 04:01, a panel of BCL lines, including 9003 (HLA-DRB1*01:01; HLA-DPB1*02:01, 13:01), 9022 (HLA-DRB1*03:01; HLA-DPB1*03:01), 9040 (HLA-DRB1*11:02; HLA-DPB1*03:01), 9053 (HLA-DRB1*13:02; HLA-DPB1*04:01) and 9080 (HLA-DRB1*01:01; HLA-DPB1*04:01, 04:02) sharing one or two of the alleles with autologous BCLs were grown in RF10 and used to stimulate the bulk CD4+ T cells (Figure 3B). Clearly, the CD4+ T cells were stimulated by 9003, 9022, 9040 and 9080, but not 9053, to secrete TNF-α and IL-2 (Figure 3C), and blocking of HLA-DR molecules on 9003 and 9080 and HLA-DP molecules on 9022 and 9040 by antibodies almost entirely hindered their ability to stimulate T cell cytokine production (Figure 3D). These together suggest that the bulk CD4+ T cell responses are HLA-DRB1*01:01- and HLA-DPB1*03:01-restricted.
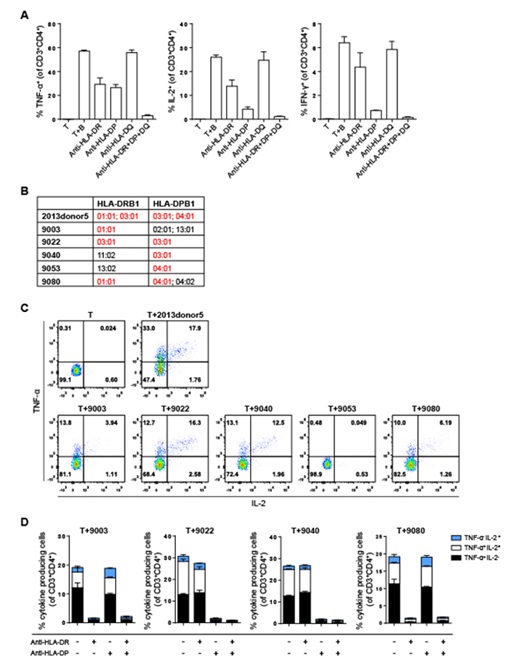 Figure 3: The response of CD4+ T cells to BSA is restricted to HLA-DRB1*01:01 and HLA-DPB1*03:01. (A)Percentages of TNF-α+, IL-2+ and IFN-γ+ cells among unstimulated and stimulated CD3+CD4+ T cells. CD3+CD4+ T cells were stimulated by treated or anti-HLA-DR, anti-HLA-DP antibody-treated, FCS-exposed autologous BCLs. (B) HLA-DR, DP molecules expressed on autologous BCLs (2013donor5) and the other five allogenic BCL lines (9003, 9022, 9040, 9053, 9080). (C) Representative FACS plots of TNF-α and IL-2 production in CD3+CD4+ T cells after stimulation by autologous and allogenic BCLs. (D) Percentages of cytokine-producing cells (TNF-α and IL-2 single and double positive cells) among stimulated CD3+CD4+ T cells.
Figure 3: The response of CD4+ T cells to BSA is restricted to HLA-DRB1*01:01 and HLA-DPB1*03:01. (A)Percentages of TNF-α+, IL-2+ and IFN-γ+ cells among unstimulated and stimulated CD3+CD4+ T cells. CD3+CD4+ T cells were stimulated by treated or anti-HLA-DR, anti-HLA-DP antibody-treated, FCS-exposed autologous BCLs. (B) HLA-DR, DP molecules expressed on autologous BCLs (2013donor5) and the other five allogenic BCL lines (9003, 9022, 9040, 9053, 9080). (C) Representative FACS plots of TNF-α and IL-2 production in CD3+CD4+ T cells after stimulation by autologous and allogenic BCLs. (D) Percentages of cytokine-producing cells (TNF-α and IL-2 single and double positive cells) among stimulated CD3+CD4+ T cells.
To narrow down the polyclonal bulk CD4+ T cell population to perhaps a more homogenous one, a TCR Vβ repertoire kit Beckman Coulter Life Sciences, USA was used to assess the bulk T cell population, as shown in Figure 4A. The larger Vβ populations including Vβ16, Vβ1 and Vβ14 were then isolated via cell sorting, stimulated by FCS-exposed autologous BCLs and intracellularly stained for TNF-α and IL-2. Compared to the bulk CD4+ T cell response, Vβ1 and Vβ14, but not Vβ16, were capable of responding to autologous BCLs in an equal magnitude (Figure 4B). We therefore concentrated on the Vβ1 population, which showed the most abundant antigen specific T cells (Figures 4A-4B). Strikingly, the CD4+ T cells expressing Vβ1 were stimulated by 9003 and 9080 but not 9022 to produce cytokines (Supplementary figure 4). This result indicates that these CD4+ T cells are only restricted to HLA-DRB1*01:01.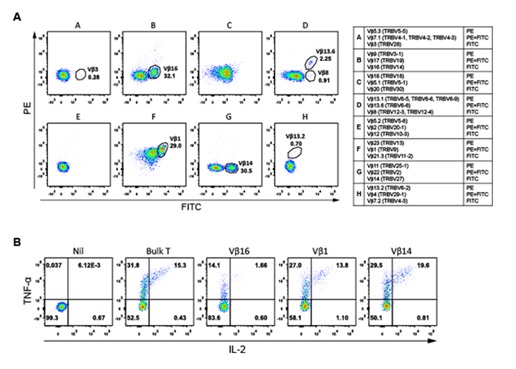 Figure 4: FCS-exposed autologous BCLs stimulate polyclonal expansion of CD4+ T cells. (A) FACS plots of TCR Vβ populations in the resting bulk CD4+ T cells. (B) TNF-α and IL-2 production in the unstimulated bulk CD4+ T cells (Nil) and the stimulated bulk CD4+ T cells and Vβ1, Vβ14 and Vβ16 populations.
Figure 4: FCS-exposed autologous BCLs stimulate polyclonal expansion of CD4+ T cells. (A) FACS plots of TCR Vβ populations in the resting bulk CD4+ T cells. (B) TNF-α and IL-2 production in the unstimulated bulk CD4+ T cells (Nil) and the stimulated bulk CD4+ T cells and Vβ1, Vβ14 and Vβ16 populations.
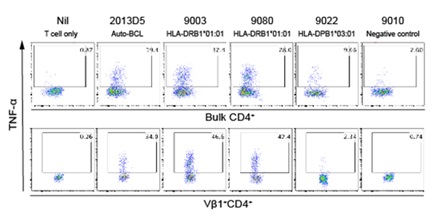 Supplementary Figure 4: The Vβ1+CD4+T cell response to BSA is restricted by HLA-DRB1*01:01. FACS plots show the production of TNF-α in unstimulated (Nil) or stimulated bulk CD4+ T cells and the Vβ1+CD4+ T cells. The T cells were stimulated by FCS-exposed autologous BCLs (2013D5) or FCS-exposed allogenic BCLs that express either HLA-DRB1*01:01 (9003, 9080) or HLA-DPB1*03:01 (9022), or neither molecule (9010).
Supplementary Figure 4: The Vβ1+CD4+T cell response to BSA is restricted by HLA-DRB1*01:01. FACS plots show the production of TNF-α in unstimulated (Nil) or stimulated bulk CD4+ T cells and the Vβ1+CD4+ T cells. The T cells were stimulated by FCS-exposed autologous BCLs (2013D5) or FCS-exposed allogenic BCLs that express either HLA-DRB1*01:01 (9003, 9080) or HLA-DPB1*03:01 (9022), or neither molecule (9010).
Identification of BSA-derived HLA-DRB1*01:01-restricted CD4+ T cell epitopes
Next, we focused on identifying the HLA-DRB1*01:01-restricted BSA epitope(s). A panel of 101 synthetic 18-mer BSA peptides (Supplementary table 1), overlapping by 12 aa, were pulsed individually onto AIM-V-grown DRB1*01:01-expressing 9003 BCLs. Peptide-pulsed 9003 APCs were then used to stimulate the bulk CD4+ T cells. Of the 101 screened peptides, BP27 (BSA157-174, FWGKYLYEIARRHPYFYA) stimulated a major TNF-α+ response, with a smaller response being elicited by BP2 (BSA7-24, ISLLLLFSSAYSRGVFRR) (Figure 5A), with a similar profile confirmed by the isolated Vβ1+ CD4+ T cells (Figure 5B), and the response of Vβ1 T cells to BSA was titratable (Figure 5C). To exclude the possibility that long-term culture might in some way bias some T cell populations, we isolated Vβ1+ CD4+ T cells from the autologous PBMCs ex vivo, labelled with CFSE and stimulated them with peptide-pulsed autologous PBMCs. Our data demonstrated that the BP27- and BP2-specific CD4+ T cells were easily detected as dominant and subdominant responses among the Vβ1 population, respectively (Figure 5D), and their response to BSA was titratable (Figure 5E). Collectively, we identified two BSA 18-mer peptides likely containing one dominant and one subdominant HLA-DRB1*01:01-restricted CD4+ T cell epitopes.
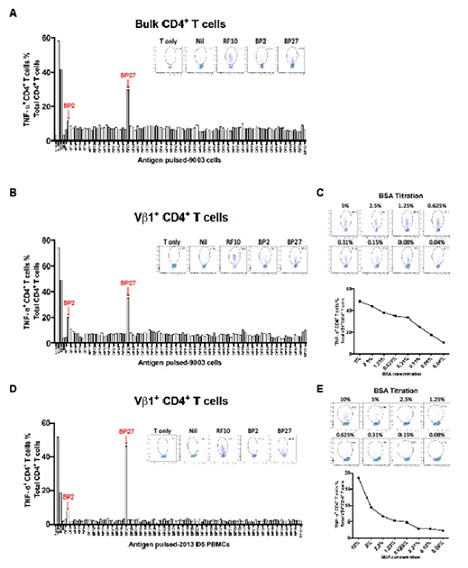 Figure 5: Identification of BSA-specific HLA-DRB1*01:01-restricted CD4+ T cell epitopes. (A, B and D) CD4+ T cell response to synthetic BSA overlapping peptides. Bar graphs show the percentage of TNF-α+ cells among bulk CD4+ T cells (A) or Vβ1+ CD4+ T cells (B) after stimulation by individual peptide-pulsed allogenic BCLs (9003), or among CFSE-labelled Vβ1+ CD4+ T cells after stimulation by peptide-pulsed autologous (2013Donor5) PBMCs (D). FACS plots show TNF-α producing cells in unstimulated T cells (T only), AIM-V medium-grown BCL-stimulated (Nil), FCS-exposed BCL-stimulated (RF10), or peptide-pulsed AIM-V medium-grown BCL-stimulated (BP2 and BP27). (C and E) TNF-α production by Vβ1+ CD4+ T cells in response to 9003 BCLs (C) or autologous PBMCs (E). 9003 BCLs (C) and autologous PBMCs (E) were cultured overnight in AIM-V media containing two-fold diluted BSA before being used to stimulate the T cells. FACS plots show TNF-α expression in the T cells. Line graphs show the percentages of TNF-α+ cells among total CD4+ T cells.
Figure 5: Identification of BSA-specific HLA-DRB1*01:01-restricted CD4+ T cell epitopes. (A, B and D) CD4+ T cell response to synthetic BSA overlapping peptides. Bar graphs show the percentage of TNF-α+ cells among bulk CD4+ T cells (A) or Vβ1+ CD4+ T cells (B) after stimulation by individual peptide-pulsed allogenic BCLs (9003), or among CFSE-labelled Vβ1+ CD4+ T cells after stimulation by peptide-pulsed autologous (2013Donor5) PBMCs (D). FACS plots show TNF-α producing cells in unstimulated T cells (T only), AIM-V medium-grown BCL-stimulated (Nil), FCS-exposed BCL-stimulated (RF10), or peptide-pulsed AIM-V medium-grown BCL-stimulated (BP2 and BP27). (C and E) TNF-α production by Vβ1+ CD4+ T cells in response to 9003 BCLs (C) or autologous PBMCs (E). 9003 BCLs (C) and autologous PBMCs (E) were cultured overnight in AIM-V media containing two-fold diluted BSA before being used to stimulate the T cells. FACS plots show TNF-α expression in the T cells. Line graphs show the percentages of TNF-α+ cells among total CD4+ T cells.
We next sought to predict the minimum epitope within BP27. Alignment of BSA and HSA aa sequences showed that BP27 shared 16 aa (FWGKYLYEIARRHPYFYA (BSA) and FKLKYLYEIARRHPYFYA (HSA)) (Supplementary figure 5 and supplementary table 2A). As HSA failed to activate our T cell line, we therefore believe W and G at positions 158 and 159 of BSA are the critical part of the epitope. Further, as the two adjacent peptides BP26 (BSA151-168) and BP28 (BSA163-180) also failed to activate the T cell line, we therefore believe only the BP27 contains the minimum epitope (Supplementary table 2B). Interestingly, the online epitope prediction tool: http://www.syfpeithi.de/index.html predicted epitopes #39 (BSA159-173, GKYLYEIARRHPYFY) and #59 (BSA158-172, WGKYLYEIARRHPYF) within the BP27 sequence (Supplementary table 2C). Based on the above results, we believe BSA158-172 is the most likely minimum epitope, as W is a preferred amino- -anchor residue (Supplementary table 2D). Similarly, #35 (BSA7-21 ISLLLLFSSAYSRGV) is the only predicted epitope within BP2 (Supplementary table 2C).
Bovine and human serum albumin amino acid Sequence alignment
 Supplementary Figure 5: Alignment of BSA and HSA aa sequences. BSA and HSA aa sequences (ALB-Bovine and ALB-human) were collected from protein database at NCBI. The GeneID was 280717 for BSA, and 213 for HSA.
Supplementary Figure 5: Alignment of BSA and HSA aa sequences. BSA and HSA aa sequences (ALB-Bovine and ALB-human) were collected from protein database at NCBI. The GeneID was 280717 for BSA, and 213 for HSA.
- An * (asterisk) indicates positions which have a single, fully conserved residue.
- A: (colon) indicates conservation between groups of strongly similar properties -scoring > 0.5 in the Gonnet PAM 250 matrix.
- A. (period) indicates conservation between groups of weakly similar properties - scoring ≤ 0.5 in the Gonnet PAM 250 matrix.
|
Peptide No. |
Amino acid location in BSA |
Synthesized BSA peptide Sequence |
Peptide No. |
Amino acid location in BSA |
Synthesized BSA peptide Sequence |
|
1 |
1-18 |
MKWVTFISLLLLFSSAYS |
52 |
307-324 |
LEKSHCIAEVEKDAIPEN |
|
2 |
7-24 |
ISLLLLFSSAYSRGVFRR |
53 |
313-330 |
IAEVEKDAIPENLPPLTA |
|
3 |
13-30 |
FSSAYSRGVFRRDTHKSE |
54 |
319-336 |
DAIPENLPPLTADFAEDK |
|
4 |
19-36 |
RGVFRRDTHKSEIAHRFK |
55 |
325-342 |
LPPLTADFAEDKDVCKNY |
|
5 |
25-42 |
DTHKSEIAHRFKDLGEEH |
56 |
331-348 |
DFAEDKDVCKNYQEAKDA |
|
6 |
31-48 |
IAHRFKDLGEEHFKGLVL |
57 |
337-354 |
DVCKNYQEAKDAFLGSFL |
|
7 |
37-54 |
DLGEEHFKGLVLIAFSQY |
58 |
343-360 |
QEAKDAFLGSFLYEYSRR |
|
8 |
43-60 |
FKGLVLIAFSQYLQQCPF |
59 |
349-366 |
FLGSFLYEYSRRHPEYAV |
|
9 |
49-66 |
IAFSQYLQQCPFDEHVKL |
60 |
355-372 |
YEYSRRHPEYAVSVLLRL |
|
10 |
55-72 |
LQQCPFDEHVKLVNELTE |
61 |
361-378 |
HPEYAVSVLLRLAKEYEA |
|
11 |
61-78 |
DEHVKLVNELTEFAKTCV |
62 |
367-384 |
SVLLRLAKEYEATLEECC |
|
12 |
67-84 |
VNELTEFAKTCVADESHA |
63 |
373-390 |
AKEYEATLEECCAKDDPH |
|
13 |
73-90 |
FAKTCVADESHAGCEKSL |
64 |
379-396 |
TLEECCAKDDPHACYSTV |
|
14 |
79-96 |
ADESHAGCEKSLHTLFGD |
65 |
385-402 |
AKDDPHACYSTVFDKLKH |
|
15 |
85-102 |
GCEKSLHTLFGDELCKVA |
66 |
391-408 |
ACYSTVFDKLKHLVDEPQ |
|
16 |
91-108 |
HTLFGDELCKVASLRETY |
67 |
397-414 |
FDKLKHLVDEPQNLIKQN |
|
17 |
97-114 |
ELCKVASLRETYGDMADC |
68 |
403-420 |
LVDEPQNLIKQNCDQFEK |
|
18 |
103-120 |
SLRETYGDMADCCEKQEP |
69 |
409-426 |
NLIKQNCDQFEKLGEYGF |
|
19 |
109-126 |
GDMADCCEKQEPERNECF |
70 |
415-432 |
CDQFEKLGEYGFQNALIV |
|
20 |
115-132 |
CEKQEPERNECFLSHKDD |
71 |
421-438 |
LGEYGFQNALIVRYTRKV |
|
21 |
121-138 |
ERNECFLSHKDDSPDLPK |
72 |
427-444 |
QNALIVRYTRKVPQVSTP |
|
22 |
127-144 |
LSHKDDSPDLPKL-KPDPN |
73 |
433-450 |
RYTRKVPQVSTPTLVEVS |
|
23 |
133-150 |
SPDLPKL-KPDPNTLCDEF |
74 |
439-456 |
PQVSTPTLVEVSRSLGKV |
|
24 |
139-156 |
L-KPDPNTLCDEFKADEKK |
75 |
445-462 |
TLVEVSRSLGKVGTRCCT |
|
25 |
145-162 |
TLCDEFKADEKKFWGKYL |
76 |
451-468 |
RSLGKVGTRCCTKPESER |
|
26 |
151-168 |
KADEKKFWGKYLYEIARR |
77 |
457-474 |
GTRCCTKPESERMPCTED |
|
27 |
157-174 |
FWGKYLYEIARRHPYFYA |
78 |
463-480 |
KPESERMPCTEDYLSLIL |
|
28 |
163-180 |
YEIARRHPYFYAPELLYY |
79 |
469-486 |
MPCTEDYLSLILNRLCVL |
|
29 |
169-186 |
HPYFYAPELLYYANKYNG |
80 |
475-492 |
YLSLILNRLCVLHEKTPV |
|
30 |
175-192 |
PELLYYANKYNGVFQECC |
81 |
481-498 |
NRLCVLHEKTPVSEKVTK |
|
31 |
181-198 |
ANKYNGVFQECCQAEDKG |
82 |
487-504 |
HEKTPVSEKVTKCCTESL |
|
32 |
187-204 |
VFQECCQAEDKGACLLPK |
83 |
493-510 |
SEKVTKCCTESLVNRRPC |
|
33 |
193-210 |
QAEDKGACLLPKIETMRE |
84 |
499-516 |
CCTESLVNRRPCFSALTP |
|
34 |
199-216 |
ACLLPKIETMREKVLASS |
85 |
505-522 |
VNRRPCFSALTPDETYVP |
|
35 |
205-222 |
IETMREKVLASSARQRLR |
86 |
511-528 |
FSALTPDETYVPKAFDEK |
|
36 |
211-228 |
KVLASSARQRLRCASIQK |
87 |
517-534 |
DETYVPKAFDEKLFTFHA |
|
37 |
217-234 |
ARQRLRCASIQKFGERAL |
88 |
523-540 |
KAFDEKLFTFHADICTLP |
|
38 |
223-240 |
CASIQKFGERALKAWSVA |
89 |
529-546 |
LFTFHADICTLPDTEKQI |
|
39 |
229-246 |
FGERALKAWSVARLSQKF |
90 |
535-552 |
DICTLPDTEKQIKKQTAL |
|
40 |
235-252 |
KAWSVARLSQKFPKAEFV |
91 |
541-558 |
DTEKQIKKQTALVELLKH |
|
41 |
241-258 |
RLSQKFPKAEFVEVTKLV |
92 |
547-564 |
KKQTALVELLKHKPKATE |
|
42 |
247-264 |
PKAEFVEVTKLVTDLTKV |
93 |
553-570 |
VELLKHKPKATEEQLKTV |
|
43 |
253-270 |
EVTKLVTDLTKVHKECCH |
94 |
559-576 |
KPKATEEQLKTVMENFVA |
|
44 |
259-276 |
TDLTKVHKECCHGDLLEC |
95 |
565-582 |
EQLKTVMENFVAFVDKCC |
|
45 |
265-282 |
HKECCHGDLLECADDRAD |
96 |
571-588 |
MENFVAFVDKCCAADDKE |
|
46 |
271-288 |
GDLLECADDRADLAKYIC |
97 |
577-594 |
FVDKCCAADDKEACFAVE |
|
47 |
277-294 |
ADDRADLAKYICDNQDTI |
98 |
583-600 |
AADDKEACFAVEGPKLVV |
|
48 |
283-300 |
LAKYICDNQDTISSKLKE |
99 |
589-606 |
ACFAVEGPKLVVSTQTAL |
|
49 |
289-306 |
DNQDTISSKLKECCDKPL |
100 |
595-607 |
GPKLVVSTQTALA |
|
50 |
295-312 |
SSKLKECCDKPLLEKSHC |
101 |
590-607 |
CFAVEGPKLVVSTQTALA |
|
51 |
301-318 |
CCDKPLLEKSHCIAEVEK |
|
|
|
Supplementary Table 1: Synthetic 18-mer BSA peptides.
A.Comparison of synthesized BP27 sequences with corresponding sequences in HSA
|
BP27 (BSA157-174) |
FWGKYLYEIARRHPYFYA |
|
HSA aa sequence |
FLKKYLYEIARRHPYFYA |
B. Synthesized BSA peptides BP26, BP27 and BP28
|
Peptide No. |
Amino acid location |
Synthesized BSA peptide Sequence |
|
BP26 |
151-168 |
KADEKKFWGKYLYEIARR |
|
BP27 |
157-174 |
FWGKYLYEIARRHPYFYA |
|
BP28 |
163-180 |
YEIARRHPYFYAPELLYY |
C. Synthesized BP27 and the predicted HLA-DRB1*0101 restricted BSA epitopes
|
Peptide No. |
Position in BSA |
Synthesized BSA peptide Sequence |
|
BP27 |
157-174 |
FWGKYLYEIARRHPYFYA |
|
#39 |
159-173 |
GKYLYEIARRHPYFY |
|
#59 |
158-172 |
WGKYLYEIARRHPYF |
|
|
|
|
|
BP2 |
7-24 |
ISLLLLFSSAYSRGVFRR |
|
#35 |
7-21 |
ISLLLLFSSAYSRGV |
D. HLA-DRB1*0101 anchors or auxiliary anchors
|
Position |
||||||||
|
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
|
Y |
|
|
L |
|
A |
|
|
L |
|
V |
|
|
A |
|
G |
|
|
A |
|
L |
|
|
I |
|
S |
|
|
I |
|
F |
|
|
V |
|
T |
|
|
V |
|
I |
|
|
M |
|
C |
|
|
N |
|
A |
|
|
N |
|
P |
|
|
F |
|
M |
|
|
Q |
|
|
|
|
Y |
|
W |
|
|
|
|
|
|
|
|
Supplementary Table 2: (A) Comparison of synthesized BP27 sequences with corresponding sequences in HAS, (B) Synthesized BSA peptides BP26, BP27 and BP28, (C) Synthesized BP27 and the predicted HLA-DRB1*0101 restricted BSA epitopes,(D) HLA-DRB1*0101 anchors or auxiliary anchors.
Discussion
With our established expertise in the expansion and culture of antigen-specific T cells, we found here that BCLs cultured in FCS-containing media alone were surprisingly capable of stimulating the activation and expansion of autologous CD4+ T cells isolated from human PBMCs. This prompted us to identify the antigens recognized by the CD4+ T cells. Our data demonstrate that it was antigens derived from an FCS component, BSA. Applying our established protocol in the identification of CD4+ T cell epitopes, we identified one dominant and one subdominant HLA-DRB1*01:01-restricted CD4+ T cell epitopes of BSA.
To our knowledge, this is the first evidence of human CD4+ T cell response to naturally processed and presented FCS protein-, BSA-, derived antigens. This is also the first report of prediction of the minimal CD4+ T cell epitopes of BSA. FCS-specific T cells have been reported for mouse [21, 22]. However, no FCS-derived antigen has been identified in the studies. A previous study did report that dendritic cells (DCs) transduced with lentiviruses in FCS-containing media stimulated autologous CD4+ T cell response to FCS [23]. However, the antigen(s) was not identified. Unlike this study, we demonstrated that the FCS-specific CD4+ T cells recognised BSA in a dose-dependent fashion and was progressively diminished when APCs were cultured in serum/BSA free medium, AIM-V. We further confirmed that human serum was not recognised by these CD4+ T cells and also excluded the possibility of EBV-antigen specificity. Ultimately, using overlapping peptides covering the full BSA sequence, we were able to identify two HLA-DRB1*01:01-restricted BSA epitopes recognized by the CD4+ T cells. Stimulation of bulk CD4+ T cells with synthetic overlapping BSA peptides identified a dominant response to BSA157-174. The response of the bulk CD4+ T cells to BSA7-24 at this stage was small. Considering multiple TCRVβ was used by the bulk CD4+ T cells, we therefore enriched the Vβ1 response, which clearly contained an easily detectable subdominant CD4+ T cell population specific to BSA7-24. Importantly, this immunodominance hierarchy was validated subsequently using ex vivo autologous PBMCs, indicating our in vitro T cell expansion did not lead to biased outcome for T cells specific to the two BSA epitopes. The two CD4+ T cell epitopes of BSA especially the immunodominant BSA157-174 we identified here could potentially be considered for epitope-based vaccines against beef allergy. However, our findings are so far limited to DRB1*01:01-restricted epitopes. It is possible that there are some more immunodominant T cell epitopes of BSA that are restricted by other HLA-I/II molecules, such as the one restricted to HLA-DPB1*03:01 shown in this study, and could potentially account for beef allergy, therefore they are expected to be identified in the future.
Intracellular cytokine staining (ICS) has been the standard approach for detecting antigen-specific T cells after stimulation [24, 25]. IFN-γ produced by activated antigen-specific T cells is the most common cytokine to be detected [15, 17, 26, 27]. However, our data show that the activated CD4+ T cells favoured in the production of TNF-α to IL-2 and IFN-γ, with a cytokine hierarchy of TNF-α > IL-2 > IFN-γ, which is quite different from the cytokine hierarchy IFN-γ > TNF-α > IL-2 identified in mouse CD8+ T cells [17], IFN-γ > IL-2 > TNF-α in human CD8+ T cells and IL-2 > IFN-γ > TNF-α in human CD4+ T cells [28-36] 5-6 h after stimulation. This raises the possibility that some T cells or T cell populations might produce cytokines with different patterns. In our cases, it might be possible that some of the T cells specific to the BSA157-174 could also weakly recognise the HSA counterpart peptide complexed with DR*01:01. It might be possible such partial activation might switch cytokine production profile. Importantly, our observation cautions the cytokine selection for detecting antigen-specific T cells. In some cases, using TNF-α as a readout cytokine may be more accurate.
In summary, this study provides the first evidence that CD4+ T cells isolated from human PBMCs can be activated and expanded by antigens derived from FCS protein BSA in cell culture. Presentation of BSA antigens to the CD4+ T cells are HLA-DRB1*01:01- and HLA-DPB1*03:01-restricted, and two HLA-DRB1*01:01-restricted BSA epitopes are identified. We therefore believe other CD4+ T cell epitopes from BSA could be presented by other HLA-II alleles, not to mention other FCS-derived antigens. This could be a matter of concern when FCS is used in human T cell cultures using PBMCs either during immune monitoring of clinical trial outcomes or direct use for immunotherapy.
Acknowledgments
This project was supported by the NHMRC program grant 567122 to WC.
References
- June CH (2007) Principles of adoptive T cell cancer therapy. J Clin Invest, 117: 1204-1212.
- Tay RE, Richardson EK,Toh HC (2021) Revisiting the role of CD4+ T cells in cancer immunotherapy-new insights into old paradigms. Cancer Gene Ther 28: 5-17.
- Cachot A, Bilous M, Liu YC, Li X, Saillard M, et al. (2021) Tumor-specific cytolytic CD4 T cells mediate immunity against human cancer. Sci Adv 7.
- Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, et al. (2008) Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 358: 2698-2703.
- Dominguez-Villar M, Hafler DA (2018) Regulatory T cells in autoimmune disease. Nat Immunol 19: 665-673.
- Zhang D, Tu E, Kasagi S, Zanvit P, Chen Q, et al. (2015) Manipulating regulatory T cells: a promising strategy to treat autoimmunity. Immunotherapy 7: 1201-1211.
- Canderan G, Gruarin P, Montagna D, Fontana R, Campi G, et al. (2010) An efficient strategy to induce and maintain in vitro human T cells specific for autologous non-small cell lung carcinoma. PLoS One 5: e12014.
- Kayser S, Bobeta C, Feucht J, Witte KE, Scheu A, et al. (2015) Rapid generation of NY-ESO-1-specific CD4+ THELPER1 cells for adoptive T-cell therapy. Oncoimmunology 4: e1002723.
- Montagna D, Maccario R, Locatelli F, Rosti V, Yang Y, et al. (2001) Ex vivo priming for long-term maintenance of antileukemia human cytotoxic T cells suggests a general procedure for adoptive immunotherapy. Blood 98: 3359-3366.
- Riddell SR, Greenberg PD (1990) The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods 128: 189-201.
- Tsai V, Southwood S, Sidney J, Sakaguchi K, Kawakami Y, et al. (1997) Identification of subdominant CTL epitopes of the GP100 melanoma-associated tumor antigen by primary in vitro immunization with peptide-pulsed dendritic cells. J Immunol 158: 1796-1802.
- Yee C, Savage PA, Lee PP, Davis MM, Greenberg PD (1999) Isolation of high avidity melanoma-reactive CTL from heterogeneous populations using peptide-MHC tetramers. J Immunol 162: 2227-2234.
- Yuwen H, Cox JH, Yewdell JW, Bennink JR, Moss B (1993) Nuclear localization of a double-stranded RNA-binding protein encoded by the vaccinia virus E3L gene. Virology 195: 732-744.
- Khanna R, Burrows SR, Kurilla MG, Jacob CA, Misko IS, et al. (1992) Localization of Epstein-Barr virus cytotoxic T cell epitopes using recombinant vaccinia: implications for vaccine development. J Exp Med 176: 169-176.
- Chen L, Zanker D, Xiao K, Wu C, Zou Q, et al. (2014) Immunodominant CD4+ T-cell responses to influenza A virus in healthy individuals focus on matrix 1 and nucleoprotein. J Virol 88: 11760-11773.
- Itoh Y, Germain RN (1997) Single cell analysis reveals regulated hierarchical T cell antigen receptor signaling thresholds and intraclonal heterogeneity for individual cytokine responses of CD4+ T cells. J Exp Med 186: 757-766.
- Gruta NLL, Turner SJ, Doherty PC (2004) Hierarchies in cytokine expression profiles for acute and resolving influenza virus-specific CD8+ T cell responses: correlation of cytokine profile and TCR avidity. J Immunol 172: 5553-5560.
- Peters T (1985) Serum albumin. Adv Protein Chem 37: 161-245.
- Paludan C, Schmid D, Landthaler M, Vockerodt M, Kube D, et al. (2005) Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 307: 593-596.
- Young LS, Dawson CW, Eliopoulos AG (2000) The expression and function of Epstein-Barr virus encoded latent genes. Mol Pathol 53: 238-247.
- Kadri N, Potiron N, Ouary M, Jegou D, Gouin E, et al. (2007) Fetal calf serum-primed dendritic cells induce a strong anti-fetal calf serum immune response and diabetes protection in the non-obese diabetic mouse. Immunol Lett 108: 129-136.
- Roner S, Zinser E, Menges M, Wiethe C, Littmann L, et al. (2008) Minor role of bystander tolerance to fetal calf serum in a peptide-specific dendritic cell vaccine model against autoimmunity: comparison with serum-free cultures. J Immunother 31: 656-664.
- Bao L, Guo H, Huang X, Tammana S, Wong M, et al. (2009) High-titer lentiviral vectors stimulate fetal calf serum-specific human CD4 T-cell responses: implications in human gene therapy. Gene Ther 16: 788-795.
- Klenerman P, Cerundolo V, Dunbar PR (2002) Tracking T cells with tetramers: new tales from new tools. Nat Rev Immunol 2: 263-272.
- Ogg GS, Mcmichael AJ (1998) HLA-peptide tetrameric complexes. Curr Opin Immunol 10: 393-396.
- Sester U, Fousse M, Dirks J, Mack U, Prasse A, et al. (2011) Whole-blood flow-cytometric analysis of antigen-specific CD4 T-cell cytokine profiles distinguishes active tuberculosis from non-active states. PLoS One 6: e17813.
- Shacklett BL, Yang O, Hausner MA, Elliott J, Hultin L, et al. (2003) Optimization of methods to assess human mucosal T-cell responses to HIV infection. J Immunol Methods 279: 17-31.
- Han Q, Bagheri N, Bradshaw EM, Hafler DA, Lauffenburger DA, et al. (2012) Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci USA 109: 1607-1612.
- Mascher B, Schlenke P, Seyfarth M (1999) Expression and kinetics of cytokines determined by intracellular staining using flow cytometry. J Immunol Methods 223: 115-121.
- Cebon J, Knights A, Ebert L, Jackson H, Chen W (2010) Evaluation of cellular immune responses in cancer vaccine recipients: lessons from NY-ESO-1. Expert Rev Vaccines 9: 617-629.
- Coulie PG, Connerotte T (2005) Human tumor-specific T lymphocytes: does function matter more than number?. Curr Opin Immunol 17: 320-325.
- Hartmann FJ, Babdor J, Gherardini PF, Amir ED, Jones K, et al. (2019) Comprehensive Immune Monitoring of Clinical Trials to Advance Human Immunotherapy. Cell Rep 28: 819-831 e814.
- Lim KP, Zainal NS (2021) Monitoring T Cells Responses Mounted by Therapeutic Cancer Vaccines. Front Mol Biosci 8: 623475.
- Lonchay C, Van Der Bruggen P, Connerotte T, Hanagiri T, Coulie P, et al. (2004) Correlation between tumor regression and T cell responses in melanoma patients vaccinated with a MAGE antigen. Proc Natl Acad Sci USA 101: 14631-14638.
- Lyons AB, Parish CR (1994) Determination of lymphocyte division by flow cytometry. J Immunol Methods 171: 131-137.
- Prendecki M, Clarke C, Edwards H, Mcintyre S, Mortimer P, et al. (2021) Humoral and T-cell responses to SARS-CoV-2 vaccination in patients receiving immunosuppression. Ann Rheum Dis 80: 1322-1329.
Citation: Lu C, Deng J, Pan J, Oveissi S, Chen W, et al. (2022) Supplemental Fetal Calf Serum in T cell Culture Media Expands Bovine Serum Albumin-Specific Human CD4+ T Cells. J Clin Immunol Immunother 8: 072.
Copyright: © 2022 Chunni Lu, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.