Treatment of Idiopathic Membranous Nephropathy (IMN)
*Corresponding Author(s):
Maria Carmen Prados SolerNephrology Department, Torrecárdenas, University Hospital, Almería, Spain
Email:lensasu@yahoo.es
Abstract
We present a 59-year-old patient with type 2 diabetes mellitus and massive nephrotic syndrome (anasarca) and biochemical syndrome. The renal biopsy showed a Membranous Nephropathy (NM). Antibodies against M-type phospholipase A2 receptor (Anti-PLA2R) positive at a very high titer. Given the existence of Idiopathic Membranous Nephropathy (IMN), treatment was started with a modified Ponticelli regimen, with no response, requiring periodic ultrafiltration sessions. Rituximab induces Nephrotic Syndrome (NS) remission in two-thirds of patients with IMN, even after other treatments have failed.We proposed treatment with rituximab based on published evidence. In IMN, the presence of M-type anti-receptor antibodies of A2 phospholipase is considered highly specific to idiopathic forms, but the presence of such antibodies has not been shown to be associated with a particular clinical profile. Assessing circulating anti-PLA2R autoantibodies and proteinuria may help in monitoring disease activity and guiding personalized rituximab therapy in nephrotic patients with IMN.
Keywords
Antibodies against M-type phospholipase A2 receptor (Anti-PLA2R); Idiopathic membranous nephropathy; Nephrotic syndrome; Rituximab
Introduction
NM is a disease characterized by the deposition of immune complexes at sub epithelial level. Its most frequent clinical presentation is NS, and it is today the first cause of NS in the Caucasian adult [1]. In recent years it has been discovered that IMN has an immunological basis. The data in favor of this alteration of the immune system are the findings found in the electron microscopy of renal biopsies, the granular deposits of immunoglobulin G (mainly IgG4) and C3 along the glomerular basement membrane, and the deposit of electrodense immunocomplexes in the sub epithelium that entails an activation of the complement products. MN may also be secondary to infections, tumors, autoimmune diseases, and use of different drugs [1]. PLA2R has been found to be the target antigen of autoantibodies in IMN patients, and is known as Anti-PLA2R. PLA2R is a type I transmembrane glycoprotein related to the animal family of type C lectin. More recently, anti-PLA2Rs have been found to be immunoglobulin’s of the IgG4 type [2].
Currently, these antibodies are present in 60-80% of IMN patients prior to immunosuppressive therapy. However, in secondary MN forms these antibodies are much less prevalent. No anti-PLA2R has been observed in other pathological conditions or in healthy individuals. In recent years, articles have been published in which several researchers have addressed the appearance of anti-PLA2r antibodies in patients with secondary MN, so more data are needed to conclude with certainty that when these antibodies are found, there is no need to investigate an underlying cause to guide a secondary MN [2,3]. Recently, various studies have described that about 70% of MNI cases are associated with the presence of anti-PLA2R. Antibody titer at diagnosis is related to the likelihood of spontaneous remission (SR) and response to treatment. However, it has not been demonstrated that in patients with MNI, the presence of anti-PLA2R antibodies is associated with a certain clinical profile of disease presentation or implies differences in clinical course, response to treatment or long-term prognosis. On the other hand, although most studies agree that the presence of anti-PLA2R antibodies is highly specific to INM, there are cases described in which the presence of these antibodies coincides with other possible etiologies and about 30% of patients with IMN are anti-PLA2R negative. In this last group of patients, antibodies have been described against other podocyte antigens whose clinical correlation is still being investigated and, therefore, there is greater uncertainty about the possible identification of secondary etiologies over time. However, since most studies have been cross-sectional, little information is available about the diagnosis of possible etiologies responsible for NM over time in both positive and negative anti-PLA2R patients [4]. In IMN the disease appears to develop by the binding of an autoantibody directed against an antigen. A podocyte that is located on the subepithelial slope of the podocyte. For this motive there are currently various immunosuppressive treatments available [2].
A very high percentage of patients (more than 40% in many series) develop spontaneous remission of the disease without any type of treatment, while another considerable percentage (around 30-40%) develops progressive renal failure accompanied by nephrotic proteinuria [2]. Ponticelli in 1989 showed that combined treatment with cytotoxics produced partial or complete remission of proteinuria in a significant proportion of patients with membranous nephropathy [5]. However, the literature is controversial regarding the effectiveness of the scheme described by Ponticelli [6]. The most important predictors of risk for a progressive decline in renal function are persistent severe proteinuria for at least three months, a reduced creatinine clearance at presentation, and a decline in creatinine clearance over the assessed proteinuria period [7].Resistant patients are defined as those with moderate or high risk disease who fail an adequate trial of treatment with both cyclophosphamide-based and calcineurin inhibitor-based regimens [8]. A trial of rituximab can be considered after a careful evaluation of the potential risks and benefits of further immunosuppression. Weak evidence suggests that a clinically relevant response to rituximab may be less likely in patients with a creatinine clearance below 75 mL/min per 1.73 m2 [9]. In this case, we present a 59-year-old patient with type 2 diabetes mellitus and massive nephrotic syndrome (anasarca) and biochemical syndrome. The renal biopsy showed a Membranous Nephropathy (NM). AntiPLA2 positive antibodies at a very high titer (366). Given the existence of IMN, treatment was started with a modified Ponticelli regimen, with no response, requiring periodic ultrafiltration sessions. We proposed treatment with rituximab based on published evidence.
Clinical Case
An 85-year-old male with a history of long-standing type 2 DM with retinopathy and diabetic neuropathy, OSAS and intrinsic asthma. Blood pressure and normal renal function. One month prior to your entry into our Service (October / 2018) refers pretibial edemas - malleolar and scrotal edema of morning onset, with worsening throughout the day and dyspnea of moderate efforts. Initially consult with your Primary Care doctor, being treated with furosemide. In the absence of a response, he is referred to the hospital. Upon admission, the patient is afebrile, dyspnoeic, conscious and oriented and presents a slight cutaneous-mucosal pallor. Blood pressure: 100/70 mmHg. Heart rate: 72 bpm. Normal head, neck without jugular vein. Rhythmic cardiac auscultation, without murmurs. Respiratory auscultation with bilateral diffuse crackles. Abdomen, without pathological findings and lower extremities with edemas ++++ / ++++ to English that leave fovea. Scrotal edema. No signs of deep vein thrombosis. Peripheral pulses preserved. Analytical on admission: leukocytes 7.30 x 10.e3 / uL with normal formula, hemoglobin 11 g / dl, hematocrit 32.5%, platelets 164 x 10.3 / uL ESR 19 mm / h, sodium 138 mM / L, potassium 3.9 mM / L, urea 110 mg / dl, creatinine 2.08 mg / dl, total cholesterol 282 mg / dl, cholesterol-HDL 88 mg / ml cholesterol-LDL 158 mg / ml, triglycerides 171 mg / dl, albumin 2.4 g / dl, total protein 4.9 g / dl. GOT, GPT, LDH, GGT, alkaline phosphatase, glucose, calcium, phosphorus and bilirubin within normal limits. Protein level in blood: albumin 44.2%, alpha-1 9.4%, alpha-2 17.4%, beta 18.2%, gamma 10.7%. In urine: proteinuria 14 g / 24 hours. Coagulation: normal, fibrinogen 676. Tumor markers: carcinoembryonic antigen, alpha-fetoprotein, PSA ng / ml and Ca 19.9: normal. ANA, ANCA, rheumatoid factor and negative glomerular baseline anti-membrane antibodies. NT-ProBNP: 200 pg / ml. Ac antiPLA2: positive (366). Kidney ultrasound showed normal-sized kidneys, with good cortico-medullary differentiation, without dilation of the urinary tract. A percutaneous renal biopsy was performed observing a renal parenchyma corresponding to the cortical zone that included 28 glomeruli. Diffuse and global thickening of the glomerular basement membranes by subepithelial deposit ("comb peaks") with mesangial focal extension not associated with mesangial cell proliferation but with flocculular-capsular adhesions. No endocapillary proliferation or glomerulitis. Absence of karyorrhexis. Irregular thickening of Bowman's capsule. Immunocomplex subepithelial deposits (IgG). Glomerular sclerosis (10 %); tubular atrophy and interstitial fibrosis (moderate); arteriosclerosis (moderate); hyaline arteriolosclerosis. Vascular changes associated with hypertension (Figures 1-6).
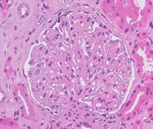 Figure 1: Staining with Hematoxylin and Eosin. 40x. Global Thickening of the capillary membranes (black arrows) of the glomerulus not accompanied by mesangial or endocapillary proliferation. Discreet thickening of Bowman's capsule (blue arrow).
Figure 1: Staining with Hematoxylin and Eosin. 40x. Global Thickening of the capillary membranes (black arrows) of the glomerulus not accompanied by mesangial or endocapillary proliferation. Discreet thickening of Bowman's capsule (blue arrow).
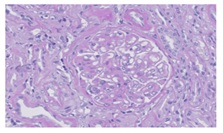 Figure 2: Pas. 40x. The technique of PAS also shows the thickening of glomerular capillary membranes (black arrows).
Figure 2: Pas. 40x. The technique of PAS also shows the thickening of glomerular capillary membranes (black arrows).
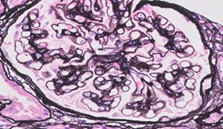 Figure 3: Jones ' Silver. 40x. subepithelial Deposits in the basal membranes of the glomerular capillaries showing an image in "Spikes de peine" (blue Arrow). This technique stains the basal membrane black. Inmuncomplejos deposits are not stained.
Figure 3: Jones ' Silver. 40x. subepithelial Deposits in the basal membranes of the glomerular capillaries showing an image in "Spikes de peine" (blue Arrow). This technique stains the basal membrane black. Inmuncomplejos deposits are not stained.
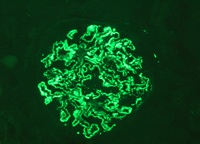 Figure 4: IgG. Subepithelial granular Deposits in capillary basal membrane with global and diffuse pattern and staining intensity 3 +/3.
Figure 4: IgG. Subepithelial granular Deposits in capillary basal membrane with global and diffuse pattern and staining intensity 3 +/3.
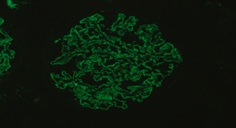 Figure 5: Lambda Light Chains. Subepithelial granular Deposits in capillary basal membrane with global and diffuse pattern and staining intensity 1-2 +/3.
Figure 5: Lambda Light Chains. Subepithelial granular Deposits in capillary basal membrane with global and diffuse pattern and staining intensity 1-2 +/3.
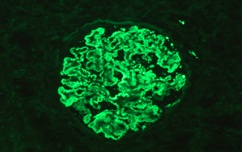 Figure 6: Kappa Light Chains. Subepithelial granular Deposits in capillary basal membrane with global and diffuse pattern and staining intensity 3 +/3.
Figure 6: Kappa Light Chains. Subepithelial granular Deposits in capillary basal membrane with global and diffuse pattern and staining intensity 3 +/3.
Depletive treatment was started with iv furosemide at high doses and iv albumin, with poor response, requiring periodic ultrafiltration sessions. Treatment was started with a modified Ponticelli regimen: prednisone at a dose of 0.5 mg / kg / day and cyclophosphamide 125 mg / day initially and then 100 mg / day, adjusted for renal function.An inhibitor of the angiotensin conversion enzyme was associated. Two months later, proteinuria has not changed. Kidney function remains normal. After two months of treatment, we have not shown changes in proteinuria. Continue to specify ultrafiltration sessions. We have decided to administer Rituximab. We will continue to monitor renal function, proteinuria and antiPLA2 Ac.
Discussion
MN is the first cause of NS in the adult [1]. From A clinical perspective, it is classified in idiopathic (IMN) or secondary depending on whether or not it is possible to identify a responsible etiology. In the absence of clinical or biochemical data indicating a specific etiology, distinguishing between the two forms can be difficult only through the data provided by the renal biopsy [2]. MN in adults is most often idiopathic (approximately 75 percent of cases) but can be caused by a variety of drugs, infections, and underlying diseases. These include gold, penicillamine, systemic lupus erythematosus, malignancy, and hepatitis B and C virus infection [2]. It is often not possible to distinguish idiopathic from secondary MN on clinical grounds alone, even though serologic studies (eg, antinuclear antibodies, hepatitis B serology) and a history of drug exposure or cancer may be revealing of a potential cause. However, there are certain findings on electron microscopy and immunofluorescence that suggest secondary disease. In patients with secondary MN, cessation of the offending drug or effective treatment of the underlying disease is usually associated with improvement in the nephrotic syndrome [10]. In view of the potential toxicity of the drugs used to treat IMN, with or without the nephrotic syndrome, the decision to initiate therapy is based, in part, upon an understanding of the natural history of untreated patients, with and without features of the nephrotic syndrome at presentation [11]:
- Spontaneous complete remission of proteinuria occurs in 5 to 30 percent at five years.
- Spontaneous partial remission (≤2 g of proteinuria per day) occurs in 25 to 40 percent at five years.
- The occurrence of end-stage renal disease in untreated patients is approximately 14 percent at five years, 35 percent at 10 years, and 41 percent at 15 years.
Risk factors for progressive idiopathic MN- In view of the often benign clinical course, immunosuppressive agents should be considered only in those with idiopathic MN who are most at risk for progressive disease or who have severe symptomatic nephrotic syndrome. Both histologic and clinical findings may be important in risk assessment. Clinical findings associated with a higher risk of developing end-stage renal disease include older age at onset (particularly greater than 50 years), male sex, nephrotic-range proteinuria (particularly if protein excretion exceeds 8 to 10 g/day), and an increased serum creatinine at presentation [12]. In contrast to these adverse risk factors, women, children, and young adults, nonnephrotic-range proteinuria, a progressive decline in protein excretion, and presentation with normal renal function have been associated with a relatively benign course [7]. In addition, patients of Asian ancestry seem to have a better long-term prognosis than other ancestries. Histologic findings are frequently regarded as important predictors of outcome, as the risk of progression is increased in patients with glomerular scarring (segmental sclerosis) and correlates more closely with the severity of the tubulointerstitial disease than with the degree of glomerular injury [12,13]. This observation is typical of most glomerular diseases. Importance of attaining remission — Attainment of a complete remission (whether spontaneous or not) is associated with good long-term outcomes. In contrast, little is known about the prognosis in patients with a partial remission [14]. A complete remission was defined as protein excretion below 0.3 g/day, while a partial remission was defined as protein excretion below 3.5 g/day plus a 50 percent or greater reduction in protein excretion from the peak value. Renal failure was defined as a creatinine clearance ≤15 mL/min, initiation of dialysis, or renal transplantation. The following findings were associated with a better renal survival on multivariate analysis that took into account clinical and laboratory data:
- Higher initial creatinine clearance and lower proteinuria at presentation.
- Lower mean arterial blood pressure over the observation period.
- Attainment of complete or partial remission in proteinuria.
Based upon this model, we define low-, moderate-, and high-risk patient subsets with varying degrees of risk for progression to more advanced kidney insufficiency (defined as a creatinine clearance ≤60 mL/min per 1.73 m2) over five years:
- Low risk: Proteinuria remains less than 4 g/day and creatinine clearance remains normal for a six-month follow-up period. Such patients have a less than 8 percent risk of developing chronic renal insufficiency over five years.
- Moderate risk: Proteinuria is between 4 and 8 g/day and persists for more than six months. Creatinine clearance is normal or near normal and remains stable over six months of observation. Chronic renal insufficiency develops over five years in approximately 50 percent of these patients.
- High risk – Proteinuria is greater than 8 g/day and persists for three months and/or renal function that is either below normal (and considered due to MN) or decreases during the observation period. Approximately 75 percent of such patients are at risk of progression to chronic renal insufficiency over five years.
Nonimmunosuppressive therapies
Given the high rate of gradual spontaneous improvement in patients with IMN, only selected patients with more severe or progressive disease should receive immunosuppressive therapy [1]. In contrast, almost all patients are candidates for more general therapies for nephrotic syndrome, such as angiotensin inhibition, lipid lowering, and, in selected patients, anticoagulation. Other aspects of therapy include diuretics to control edema and maintenance of adequate nutrition.
- Proteinuria goal: The optimal proteinuria goal in patients with chronic kidney disease is less than 1000 mg/day. However, this goal is often not attainable in patients with IMN.
- Goal blood pressure: The goal blood pressure in patients with MN is the same as it is in other patients with proteinuric chronic kidney disease (125/75 mmHg). Attainment of this goal can slow the progression of proteinuric chronic kidney disease and can provide cardiovascular protection since chronic kidney disease is associated with a marked increase in cardiovascular risk. The data supporting these recommendations are presented separately. Attainment of the blood pressure goal in patients with MN usually requires more than angiotensin inhibition alone. Correction of volume overload is of particular importance and usually requires loop diuretics. Diuretics should be pushed until the blood pressure goal is reached or the patient has attained "dry weight" which, in the presence of persistent hypertension, is defined as the weight at which further fluid removal leads to symptoms (fatigue, orthostatic hypotension) or to decreased tissue perfusion as evidenced by an otherwise unexplained elevation in the blood urea nitrogen and/or serum creatinine concentration. A low-salt diet is an important component of antihypertensive therapy (especially when using angiotensin inhibitors) and edema control in patients with MN. In addition, a high-salt diet can increase proteinuria, and in some individuals, a high-salt diet rather than increased immunologic activity should be considered as an underlying cause of worsening proteinuria.
- Lipid lowering: Hyperlipidemia, with often dramatic elevations in the serum cholesterol concentration, is commonly present in patients with nephrotic syndrome. The mainstay of therapy for such hypercholesterolemia is statins.
Immunosuppressive therapies
Indications for and choice of therapy- Since many patients with mild to moderate disease undergo spontaneous remission and immunosuppressive agents have appreciable toxicity, the decision to treat must be based upon the probability that the patient will have progressive disease (defined as an otherwise unexplained elevation in serum creatinine or persistent high-grade or increasing proteinuria in patients at moderate to high risk for progression) [15]. The treatment regimen must be based upon the risk of progressive disease. First-line immunosuppressive therapy consists of cytotoxic drugs (usually cyclophosphamide) plus glucocorticoids or a calcineurin inhibitor with low-dose or no glucocorticoids (a regimen based upon cytotoxic drugs is preferred in some high-risk patients with declining glomerular filtration rate due to MN and an estimated glomerular filtration rate above 30 mL/min/1.73 m2). Patients who do not respond to one regimen are usually treated with the other, and those with resistant disease may be treated with rituximab.
In our case, it is a patient with a high risk of progression.
High risk for progression- High-risk patients with idiopathic MN are defined as those with protein excretion exceeding 8 g/day that persists for more than three months and/or renal function that is either below normal (and considered due to MN) or decreases during the observation period, despite maximum nonimmunosuppressive therapy. These patients are also likely to have prominent nephrotic symptoms or signs, such as marked hypoalbuminemia and edema. Approximately 75 percent of such patients progress to worsened renal insufficiency over five years. Rituximab has been used in patients with idiopathic membranous nephropathy who have failed previous treatment with other immunosuppressive regimens. Rituximab may have benefit among patients with a moderate risk of progression who have not previously received immunosuppressive therapy [8, 16]. In one unblinded trial for 12 months, the rate of complete or partial remission was higher among patients treated with rituximab (65 versus 34 percent). These findings are consistent with observational studies that demonstrate a maximal reduction in proteinuria at 18 to 24 months after treatment with rituximab. Anti-PLA2R antibodies, which were present in 73 percent of patients at baseline, disappeared in a greater proportion of patients receiving rituximab (50 versus 12 percent). Serious adverse events were similar between the two groups.
Resistant disease- The optimal approach to moderate- or high-risk patients with stable renal function who fail treatment with both cyclophosphamide and calcineurin inhibitor-based regimens is not known. We prefer a trial of rituximab in such patients, although limited data are available suggesting efficacy. Several observational (nonrandomized) studies in patients with idiopathic resistant MN have reported outcomes following the administration of rituximab. Rituximab therapy is generally well tolerated, adverse effects are minor and primarily consisted of infusion reactions. AntiPLA2R-positive patients with lower titers had significantly greater remission rates compared with patients who had higher titers. Rituximab may provide benefit to patients who failed prior immunosuppressive therapy, especially those with relatively preserved renal function. Four weekly doses of rituximab (375 mg/m2) appear to have the same effect on proteinuria reduction as a regimen of 1 g every two weeks. It is suggested the somewhat simpler and cheaper regimen of a dose of 1 g given intravenously and repeated in two weeks. Patients who continue to have significant proteinuria may have this dose repeated at six months. PLA2R is a transmembrane receptor that is highly expressed in glomerular podocytes and has been identified as a major antigen in human idiopathic MN. A decline in anti-phospholipase A2 receptor (PLA2R) antibodies may predict the clinical response to rituximab treatment [17,18]. The anti-PLA2R autoantibody-negative patients may be in the midst of a spontaneous or treatment-induced remission.The monitoring serum anti-PLA2R antibodies may allow a more accurate assessment of the immunological response to rituximab (and possibly other therapies) than is provided by measurement of proteinuria alone [17, 18].
Conclusion
MN is among the most common causes of the nephrotic syndrome in nondiabetic adults, accounting for up to one-third of biopsy diagnoses. A significant percentage (15-50% of cases) of patients with IMN develop progressive chronic kidney disease. Rituximab induces NS remission in two-thirds of patients with IMN, even after other treatments have failed. The rate of complete or partial remission was higher among patients treated with Rituximab. Therefore, assessing circulating anti-PLA2R autoantibodies and proteinuria may help in monitoring disease activity and guiding personalized rituximab therapy in nephrotic patients with IMN. The monitoring serum anti-PLA2R antibodies may allow a more accurate assessment of the immunological response to rituximab (and possibly other therapies) than is provided by measurement of proteinuria alone.
References
- Fervenza FC, Sethi S, Specks U (2008) Idiopathic membranous nephropathy: diagnosis and treatment. Clin J Am Soc Nephrol 3: 905.
- Debiec H, Ronco P (2011) PLA2R autoantibodies and PLA2R glomerular deposits in membranous nephropathy. N Engl J Med 364: 689-690.
- Qin W, Beck LH Jr, Zeng C, Chen Z, Li S, et al. (2011) Anti-phospholipase A2 receptor antibody in membranous nephropathy. J Am Soc Nephrol 22:1137-1143.
- Hofstra JM, Beck LH Jr, Beck DM, Wetzels JF, Salant DJ (2011)Anti-phospholipase A2 receptor antibodies correlate with clinical status in idiopathic membranous nephropathy. Clin J Am Soc Nephrol 6: 1286-1291.
- Ponticelli C, Zucchelli P, Passerini P, et al. (1995) A 10-year follow-up of a randomized study with methylprednisolone and chlorambucil in membranous nephropathy. Kidney Int 48:1600-1604.
- Ponticelli C, Altieri P, Scolari F, Passerini P, Roccatello D, et al. (1998) A randomized study comparing methylprednisolone plus chlorambucil versus methylprednisolone plus cyclophosphamide in idiopathic membranous nephropathy. J Am Soc Nephrol 9: 444-450.
- Reichert LJ, Koene RA, Wetzels JF (1998) Prognostic factors in idiopathic membranous nephropathy. Am J Kidney Dis 31: 1-11.
- Dahan K, Debiec H, Plaisier E, Cachanado M, Rousseau A, et al. (2017) Rituximab for Severe Membranous Nephropathy: A 6-Month Trial with Extended Follow-Up. J Am Soc Nephrol 28: 348-358.
- Ruggenenti P, Cravedi P, Chianca A, Perna A, Ruggiero B, et al. (2012) Rituximab in idiopathic membranous nephropathy. J Am Soc Nephrol 23:1416-1425.
- Troyanov S, Roasio L, Pandes M, Herzenberg AM, Cattran DC, et al. (2006) Renal pathology in idiopathic membranous nephropathy: a new perspective. Kidney Int 69:1641-1648.
- Jha V, Ganguli A, Saha TK, Kohli HS, Gupta KL, et al. (2007) A randomized, controlled trial of steroids and cyclophosphamide in adults with nephrotic syndrome caused by idiopathic membranous nephropathy. J Am Soc Nephrol 18:1899-1904.
- Shiiki H, Saito T, Nishitani Y, Mitarai T, Yorioka N, et al. (2004) Prognosis and risk factors for idiopathic membranous nephropathy with nephrotic syndrome in Japan. Kidney Int 65: 1400-1407.
- Wu Q, Jinde K, Nishina M, Tanabe R, Endoh M, et al. (2001) Analysis of prognostic predictors in idiopathic membranous nephropathy. Am J Kidney Dis 37: 380-387.
- Troyanov S, Wall CA, Miller JA, Scholey JW, Cattran CL, et al. (2004) Idiopathic membranous nephropathy: definition and relevance of a partial remission. Kidney Int 66: 1199-1205.
- Philibert D, Cattran D (2008) Remission of proteinuria in primary glomerulonephritis: we know the goal but do we know the price? Nat Clin Pract Nephrol 4: 550-559.
- Ruggenenti P, Debiec H, Ruggiero B, Chianca A, Gaspari F, et al. (2015) Anti-Phospholipase A2 Receptor Antibody Titer Predicts Post-Rituximab Outcome of Membranous Nephropathy. J Am Soc Nephrol 26: 2545-2558.
- Cattran DC, Kim ED, Reich H, Hladunewich M, Kim J, et al. (2017) Membranous Nephropathy: Quantifying Remission Duration on Outcome. J Am Soc Nephrol 28: 995-1003.
- Cravedi P, Ruggenenti P, Remuzzi G (2011) Circulating anti-PLA2R autoantibodies to monitor immunological activity in membranous nephropathy. J Am Soc Nephrol 22: 1400-1402.
Citation: Maria Carmen Prados Soler, Isabel Villegas Pérez, Maria Dolores Salmerón Rodriguez, Marta Pandero Moya, Francisco Javier González Martínez (2023) Treatment of Idiopathic Membranous Nephropathy (IMN). J Nephrol Renal Ther 9: 077.
Copyright: © 2023 Maria Carmen Prados Soler, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.