Two Roads Converging: Mitochondria and Inflammatory Signaling
*Corresponding Author(s):
Luis MateronDepartment Of Biology, University Of Texas-Pan American, Texas, United States
Tel:9566657140,
Fax:9566653657
Email:lmateron@utpa.edu
Abstract
As the complexity of cellular signaling in inflammatory response emerges, it is increasingly clear that mitochondria are directly involved in, and in some cases are even required for, activation of inflammatory response. As a bioenergetic organellar network, mitochondria dynamically modulate their organization and function in response to cellular signaling cues and metabolic demand. The NLRP3 inflammasome, a caspase-activating multifactor scaffolding assembly, is directly activated by mitochondrial factors and functional parameters. Mitochondria are also heavily implicated as downstream targets of inflammation in a variety of tissues. Elevated inflammation and cytokine-mediated damage to mitochondria are implicated in the pathogenesis of disparate conditions such as Type 2 diabetes and autism spectrum disorders. Recent findings indicate that mitochondrial factors are released as extracellular mediators of inflammatory response. Here, we discuss the mechanistic interaction of mitochondria in inflammatory signaling, as well as the implications for inflammatory mitochondrial damage as a causative force in highly prevalent human diseases.
INFLAMMATORY SIGNALING AND INNATE IMMUNE RESPONSE
Inflammation is a crucial mechanism of innate immune response. Macrophages and neutrophils recognize pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), undergoing a complex set of signaling interactions to release pro-inflammatory cytokines (most notably IL-1β and IL-18 [1] that prompt inflammatory response. To mediate innate immune response, pattern-recognition receptors (PRRs) recognize a broad range of markers of infection, stress, and damage. PRRs include membrane-bound receptors such as Toll-like receptors (TLRs), the interleukin receptors (ILRs), and the tumor necrosis factor receptors 1 (TNF-R1) and 2 (TNF-R2). Upon binding of extracellular ligands, these PRRs activate intracellular signaling events, such as activation of NF?B, a transcription factor that upregulates expression of a wide variety of stress-response genes, or through post-translational modification such as activation of c-Jun amino-terminal kinase (JNK) [2] to effect inflammatory response. Within the cytoplasm, inflammatory ligands bind and activate intracellular PRRs that combine with a variety of associated factors to form large cytoplasmic scaffolding assemblies that integrate inflammatory activation and activate secretion of the major cytokines IL-1β and IL-18 by binding and activating caspase-1 [3] (Figure 1). These cytoplasmic PRRs, classed as nucleotide-binding domain leucine-rich repeat-containing receptors (NLRs), recognize a wide variety of intracellular inflammatory stimuli. Four classes of NLRs (NLRP1, NLRP3, NLRC4 and AIM2) share in common a nucleotide-binding oligomerization domain, and have demonstrated an ability to form large oligomeric inflammasome complexes in the cytoplasm [4]. While each of the four sense a variety of inflammatory signals to mediate caspase-dependent activation of inflammation, the NLRP3 inflammasome is the best characterized.
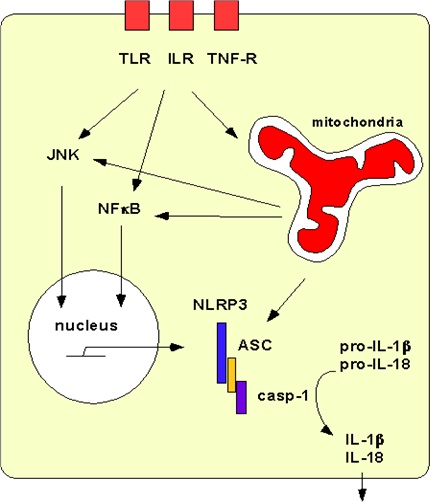
Figure 1: Schematic of mitochondrial interactions with NLRP3 inflammasome signaling.
Plasma membrane PRRs TLR, ILR, and TNF-R (red boxes) bind cytokines and extracellular ligands, activating NF?B and JNK, which activate nuclear transcription of cellular stress factors, particularly NLRP3. NLRP3 (blue), ASC (gold), and caspase-1 (purple) associate in the cytoplasm as the large, macromolecular NLRP3 inflammasome in macrophages. Mitochondria are impacted by membrane-bound PRR signals and aid in activating the NLRP3 inflammasome (via ROS or specific interaction with mtDNA, cardiolipin, MFN2, or MAVS). Inflammasome activation results in cleavage of pro-IL-1β and pro-IL-18 to active cytokines IL-1β and IL-18, which are secreted from the cell to spread inflammation.
THE NLRP3 INFLAMMASOME
The NACHT, LRR, and PYD domain-containing protein (NLRP3) is a major mediator of caspase-1 activation. To accomplish this, NLRP3 associates with the apoptosis-associated speck-like protein containing a CARD domain (ASC) adaptor protein. ASC contains a caspase recruitment domain (CARD), thus allowing binding of pro-caspase-1 to the inflammasome complex. ASC has a remarkable ability to dimerize and associate with pro-caspase-1, causing formation of a single large (~2 µm diameter) NLRP3 inflammasome in macrophages [5].
Formation of the NLRP3 inflammasome is cued by the membrane-bound PRRs, such as the TLRs, ILRs, and TNF-Rs, which activate NF?B and JNK in the nucleus NF?B is a major stress-response transcription factor, which rapidly increases mRNA levels of pro-inflammatory factors, particular NLRP3 and pro-IL-1β [6,7]. For inflammasome formation, NF?B-mediated increases in transcription of both NLRP3 and pro-IL-1β are required, as these factors are found at low basal levels. Conversely, ASC and pro-caspase-1 (as well as pro-IL-18) are found at sufficiently high levels in the cytoplasm to allow inflammasome assembly [6]. Upon binding of caspase-1 as part of the NLRP3 inflammasome, the inactive pro-caspase-1 is autocatalytically cleaved and forms the active caspase-1 heterodimer [8,9]. Active caspase-1 then cleaves proIL-1β and pro-IL-18 to their active Il-1β and IL-18 forms, which are then secreted as inflammatory cytokines [8-10] (Figure 1). An exciting collection of findings indicates that a variety of factors located in the mitochondria play a crucial role in NLRP3 inflammasome response.
MITOCHONDRIA: A DYNAMIC ORGANELLAR NETWORK
As organelles of endosymbiontic origin [11], mitochondria occupy a highly unique niche in cellular biology. By combining genetic contributions from both chromosomal and mitochondrial genomes, mitochondria carry out the bulk of cellular ATP production via oxidative phosphorylation. Mitochondrial structure is incredibly dynamic, changing in response to cellular need and organellar bioenergetic function, even in cells with highly constrained architecture such as cardiac [12] and skeletal muscle fibers [13]. This dynamic structure undergoes profound alteration in response to mitochondrial dysfunction, indicating that structural dynamics represent a critical parameter to be explored in mitochondrial-immune interactions.
The dual genetic composition of mitochondria is unique among the organelles of a human cell: both nuclear- and mitochondrially-encoded gene products are required to fully assemble the five complexes of oxidative phosphorylation (OxPhos) in the mitochondrial inner membrane. While hundreds of proteins are present in human mitochondria [14,15], the vast majority of these are encoded by nuclear genes. Mitochondrial DNA (mtDNA) encodes only 2 rRNAs, 22 tRNAs, and 13 polypeptides from a 16,569 bp circular DNA. Despite the small handful of proteins encoded by mtDNA, these polypeptides are essential subunits of the OxPhos complexes in the inner membrane. While Complex II is encoded solely by nuclear DNA, the other four complexes each contain at least one mtDNA-encoded polypeptide. Complexes I-IV transfer electrons supplied by NADH and FADH2, ultimately donated to molecular oxygen to create water, to drive H+ pumping from the mitochondrial matrix to the intermembrane space. This H+ pumping activity generates a proton-motive force, which in higher organisms is chiefly comprised of an electrochemical gradient, or transmembrane potential (??m) that is then utilized by the F1F0 ATP synthase [16]. By allowing a single H+ to return to the matrix down the gradient, ADP and Pi are bound at the F1 portion of the ATP synthase and coalesced to ATP during the rotation-mediated conformational shifting of the holoenzyme [17].
Mitochondrial structure is organized to support bioenergetic function. While traditional models of mitochondrial ultrastructure envisioned a collection of individual organelles dispersed throughout the cytoplasm, advances in cellular imaging and the identification of genetic factors controlling mitochondrial morphology have combined to reveal mitochondrial ultrastructure as a highly dynamic, sensitive process capable of dramatic response to cellular stimuli. Mitochondria were originally named as being 'thread-like granules'. Improved imaging techniques revealed that mitochondria do in fact have the ability to interconnect as a networked reticulum throughout the cell [12,18]. Optic atrophy 1 (OPA1) was identified as a factor that is required for fusion of the mitochondrial inner membrane [19], while mitofusin 1 (MFN1) and mitofusin 2 (MFN2) carry out fusion of the outer mitochondrial membrane (Hoppins et al., 2007). While OPA1, MFN1, and MFN2 carry out mitochondrial fusion, a different set of factors carry out mitochondria fission. FIS1 [20] and MFF [21] are mitochondrial outer membrane proteins that recruit dynamin-related protein 1 (DRP1) from the cytoplasm. Upon docking at the outer membrane, DRP1 will form multimeric rings around a mitochondrion, dividing it in two [23]. Thus, mitochondrial fusion and fission are opposing processes governed by different sets of factors, in which a cell will balance mitochondrial organization between the two (Figure 2).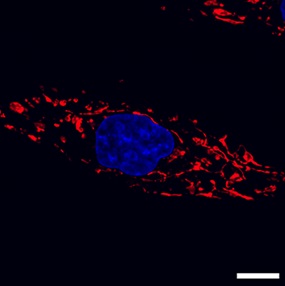
Figure 2: 3T3 mouse embryonic fibroblasts visualized by confocal fluorescence microscopy, labeled for mitochondria (MitoTracker, red) and the nucleus (DAPI, blue).
The cell's mitochondria display both interconnected, fused mitochondria (to the right of the nucleus) and fragmented, divided mitochondria (to the left of the nucleus). Size bar = 10 µm.
Mitochondrial ultrastructure is directly tied to mitochondrial function, existing in a sensitive balance to maintain energetic homeostasis. Damage or dysfunction to the structure/function balance of mitochondria causes a loss of ability to maintain a fused, interconnected mitochondrial network. Cells with either genetic [23] or pharmacologically-induced mitochondrial dysfunction [24] have mitochondria that are unable to fuse together, instead maintaining an obligately fragmented organization. This loss of organellar fusion under conditions of mitochondrial dysfunction is caused by proteolytic cleavage of the OPA1 fusion protein [25], occuring when the ??m across the mitochondrial inner membrane is low [26]. Subsequently, ??m-sensitive cleavage of OPA1 was found to be mediated by OMA1, a metalloprotease located in the inner mitochondrial membrane [27,28]. Thus, mitochondrial function (specifically ??m) directly mediates mitochondrial fusion by OPA1, while transgenic ablation of either mitochondrial fission [29] or fusion [30] negatively impacts bioenergetic function, indicating the hand-in-hand relationship between mitochondrial bioenergetic function and structural organization. Moreover, while bioenergetic dysfunction and loss of efficient mitochondrial organization are detrimental in and of themselves, emerging evidence suggests that mitochondrial dysfunction plays a strong role in activation of NLRP3-mediated inflammatory signaling (discussed below). As mitochondrial structural dynamics are directly linked to processes including apoptosis and autophagy [31,32], mitochondrial-inflammatory interactions are likely to be similarly linked to a fascinating set of dynamic alterations, with enormous consequences for the cell and surrounding environment.
MITOCHONDRIA IN NLRP3 INFLAMMASOME SIGNALING
Mitochondria are emerging as a major activator of NLRP3 inflammasome signaling, and are in some cases required for NLRP3 inflammasome activation. Recent findings show that a variety of mitochondrial components interacts with and activate the NLRP3 inflammasome as major contributors to innate immune signaling by macrophages. Even more intriguingly, Misawa et al. showed that mitochondria are recruited en masse to the inflammasome within the cytoplasm of macrophages [33], suggesting that a host of other mitochondrial factors may have critical roles in inflammasome-mediated signaling.
A role for mitochondria in NLRP3 inflammasome signaling was first suggested when mitochondria were observed to colocalize with the NLRP3/ASC/caspase-1 scaffold assembly upon inflammasome induction. While reactive oxygen species (ROS) had previously been shown to be necessary for NLRP3 inflammasome activity [34], inhibition of the mitochondrial voltage-dependent anion channel (VDAC) abrogated both intracellular ROS levels and inflammasome assembly, indicating that mitochondrial ROS production is directly involved in NLRP3 inflammasome signaling of macrophages [35]. Subsequently, macrophages treated with E. coli lipopolysaccharide or ATP (both inflammasome activators) displayed release of mtDNA into the cytoplasm. Moreover, transfection to deliver cytoplasmic mtDNA stimulated secretion of both IL-1β and IL-18, indicating that release of mtDNA from the mitochondrial matrix into the surrounding cytoplasm directly contributes to NLRP3 inflammasome activity. Shimada et al. [36] then found that mitochondrial dysfunction correlates with NLRP3 inflammasome activity, with binding of oxidized mtDNA a required step in NLRP3 inflammasome activation and IL-1β [36]. These results provide an unusual mechanism of inflammasome activation: while mtDNA damage is increasingly appearing as a common form of mitochondrial damage in a variety of cellular settings [37,38], it is unclear how a highly packaged, compacted circle of DNA [39,40] is released from a double membrane-bound organelle into the cytoplasm. Future research will undoubtedly shed new light on how mtDNA escapes the organelle to participate in inflammatory signaling.
Additional studies suggest that entire mitochondrial organelles are active players in NLRP3 inflammasome activity. Cardiolipin, a diphosphatidylglycerol lipid, is found nearly exclusively in the mitochondrial inner membrane. However, upon both ROS-dependent and -independent induction of the NLRP3 inflammasome, cardiolipin translocates to the outer membrane of the mitochondria, where it interacts with the leucine-rich repeat (LRR) domain of NLRP3, concurrent with ASC and caspase-1 recruitment to the inflammasome leading to IL-1β secretion [41]. Cardiolipin translocation to the outer membrane appears to be mediated by phospholipid scramblase-3, contributing to mitochondrial autophagy in rat cortical neurons [42], suggesting that binding of cardiolipin by cytoplasmic signaling molecules is a general stress-response mechanism in cells, with profound disease implications. As a large-scale cytoplasmic macromolecular scaffolding assembly, the NLRP3 inflammasome requires the tubulin cytoskeleton to transport mitochondria to inflammasome sites, where they bind to ASC [33]. The mitochondrial anti-viral signaling (MAVS) factor is a likely adaptor protein mediating NLRP3-mitochondrial interaction, required for IL-1β maturation and secretion in THC-1 monocytes, as well as macrophages [43,44]. MFN2 is required for NLRP3 inflammasome activation following infection with RNA virus: Ichinohe et al. found that this association requires an intact ??m for MFN2-NLRP3 interaction [45]. These studies indicate that a diverse set of mitochondrial factors mediate activation of the NLRP3 inflammasome (Figure 1). MFN2 and MAVS, as proteins located in the outer mitochondrial membrane, are easier to envision as 'docking partners' with the NLRP3 protein, while translocation of cardiolipin to the mitochondrial outer membrane and the release of mtDNA from the organelle into the cytoplasm represent highly dynamic events in inflammasome activation. These studies clearly demonstrate that much remains to be understood regarding the roles these factors play in inflammatory response, and further indicate that additional mitochondrial factors localized to any part of the organelle may be similarly involved in dynamic recruitment to the NLRP3 inflammasome. Further, the studies above (except where indicated) have characterized NLRP3 signaling interactions in macrophages, as major mediators of innate immune response. It is highly likely that NLRP3-mediated signaling will show a range of specific responses in different cell types throughout the body.
INFLAMMATORY DAMAGE TO MITOCHONDRIA
In addition to their emerging role as integral contributors to inflammatory response in the innate immune system, mitochondria are increasingly implicated as cellular targets of cytokine-mediated inflammation in a host of tissues. While macrophages and similar immune cells involve mitochondria in inflammatory signaling, many of the same cytokines have been shown to damage mitochondria in the pathogenesis of prevalent human diseases, particularly Type 2 diabetes mellitus.
Mitochondria have long been implicated in the pathogenesis of Type 2 diabetes mellitus and associated metabolic disorders. Decreased mitochondrial function [46,47] and gene expression [48,49] have been strongly correlated with Type 2 diabetes in diverse tissues such as skeletal muscle and peripheral blood. As such, an abundance of clinical and experimental data indicates that decreased mitochondrial function and bioenergetics capacity plays a contributing role in Type 2 diabetes. However, it has been unclear what genetic or environmental factors this can be attributed to. Inherited mutations of mtDNA cause insulin resistance and diabetes mellitus in patients [50,51], indicating that mitochondrial dysfunction can be a causative determinant of insulin resistance. Despite this, inherited pathogenic mtDNA mutations do not occur frequently enough (1 in 5,000-10,000 individuals [52,53] to explain the rapidly-expanding prevalence of Type 2 diabetes worldwide. However, the emergence of cytokine-mediated inflammation as a causative mechanism of Type 2 diabetes suggests that cytokine-mediated damage to mitochondria plays a major role in the pathogenesis of diabetes and co-morbid conditions.
Cytokine-mediated inflammation has gained recognition as a major causative force in the development of insulin resistance and diabetes [54-56]. While the initial studies demonstrating this causative mechanism explored the ability of TNF-a to mediate insulin resistance [55], subsequent studies built upon these findings to include IL-1β, IL-6, IL-18, resistin, leptin, adiponectin, and others. Many of these are expressed both by macrophages and adipocytes, providing a mechanistic link for the co-morbidity of Type 2 diabetes and obesity [57]. Upon binding of these cytokines to PRRs at the plasma membrane, the NF?B and JNK pathways are activated, mediating crucial stress-mediated transcription of inflammatory factors (such as IL-1β and NLRP3, above). The NLRP3 inflammasome is activated in cytokine-mediated insulin resistance [58,59] and mediates impaired wound healing through sustained inflammation in diabetic patients [60].
Many of these same cytokines have been shown to directly damage mitochondria. TNF-a cause rapid damage to mtDNA and increased ROS production [61], and inhibits mitochondrial bioenergetics [62]. This TNF-a-induced damage is dependent on TNF-R1 binding, and appears to involve stress-response translocation of p53 to mitochondria [61,63]. Similarly, heat-inactivated E. coli activates TLR-4, causing mtDNA depletion [64]. These findings are concordant with experimental and clinical data showing loss of mtDNA content and bioenergetic function [65-67]. This mitochondrial damage has wide-ranging effects on the cell at large. Mitochondrial involvement in the development of insulin resistance appears to occur via elevated mitochondrial ROS production, rather than decreased OxPhos activity per se [68]. Mitochondrial dysfunction has been suggested to cause insulin resistance by decreasing insulin receptor substrate-1 (IRS1) expression [69,70]. Mechanistically, loss of mtDNA has been shown to effect broad changes in nuclear transcription via 'retrograde' mitochondria-to-nucleus signaling, in which mitochondrial dysfunction affects pathways including NFkB and JNK to add to cellular stress response [71,72]. The specific impacts of mitochondrial dysfunction on gene expression of various inflammatory factors are likely to provide insight into a critical cell-wide consequence of mitochondrial dysfunction.
While the connections between inflammatory signaling and mitochondria have been best characterized in diabetes and metabolic disorders, these interactions are increasingly found across a range of other prevalent diseases. Gene expression profiling [73,74] and biomarker studies find increases in cytokines in autistic subjects [75], particularly IL-6 and TNF-a [76-]. These studies are consistent with findings of mitochondrial dysfunction and increased organellar fission in brain samples of autistic individuals [79]. Similar involvement of NLRP3-associated inflammation is found in Alzheimer's disease [80] and cardiomyopathy [81]. These associations strongly suggest that inflammation and mitochondria comprise a pathogenic axis that is likely to play a role in a wide range of prominent human diseases.
MITOCHONDRIAL FACTORS AS INFLAMMATORY MESSENGERS
As discussed above, the release of mtDNA from the organelle in response to NLRP3-associated inflammatory stimuli is a highly novel, dynamic response, suggesting that mtDNA is a key mediator of intracellular inflammation. Recent evidence suggests that mtDNA and associated factors may have further roles as extracellular inflammatory mediators. Mathew et al. observed that extracellular, partially degraded mtDNA causes induction of cytokine secretion, most notably IL-1β, suggesting that mtDNA is itself a category of damage-associated molecular pattern (DAMP) for recognition by PRRs [82]. Similarly, Chaung et al. [83] found that transcription factor A, mitochondrial (TFAM), the major mtDNA-packaging protein, mediates inflammation in hemorrhagic shock [83]. These findings strongly indicate that mtDNA and the other protein factors directly associated with it [39] are released from mitochondria and act as pro-inflammatory signaling factors, appearing as clinically-relevant indicators of inflammation in experimental systems and patients [84,85]. Just as the adaptive nature of the mitochondrial network has become evident as a key element of cellular homeostasis, these findings indicate the importance of mitochondrial factors as mediators of inflammatory signaling. The mechanisms of mitochondrial factor release will reveal exciting, highly novel molecular dynamics of factors previously thought to reside exclusively within mitochondria.
CONCLUSION
The specific interactions of mitochondrial factors with innate inflammatory factors, particularly NLRP3, bring together two fields of cell biology that previously had little apparent connection. These interactions are of enormous importance medically, as increased inflammation has emerged as a causative or contributory factor in a wide range of many of the most rapidly-expanding diseases today. As the dynamics and causes of both bioenergetic stress and inflammation in human disease are uncovered, understanding the intrinsic mechanistic connections of inflammatory signaling with mitochondrial biology is increasingly vital to cellular homeostasis and human health.
ACKNOWLEDGEMENTS
This work is supported by a Research Grant from the Diabetes Action Research and Education Foundation (to RG).
REFERENCES
- Garlanda C, Dinarello CA, Mantovani A (2013) The interleukin-1 family: back to the future. Immunity 39: 1003-1018.
- Nguyen MT, Satoh H, Favelyukis S, Babendure JL, Imamura T, et al. (2005) JNK and tumor necrosis factor-alpha mediate free fatty acid-induced insulin resistance in 3T3-L1 adipocytes. J Biol Chem 280: 35361-35371.
- Martinon F, Burns K, Tschopp J (2002) The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10: 417-426.
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10: 241-247.
- Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, et al. (2007) The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 14: 1590-1604.
- Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, et al. (2009) NF-kB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 183:787-791.
- Fitzgerald DC, Meade KG, McEvoy AN, Lillis L, Murphy EP, et al. (2007) Tumour Necrosis Factor-alpha (TNF-alpha) increases Nuclear Factor kappaB (NFkappaB) activity in and Interleukin-8 (IL-8) release from bovine mammary epithelial cells. Vet Immunol Immunopathol 116:59-68.
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, et al. (1992) A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356: 768-774.
- Wilson KP, Black JA, Thomson JA, Kim EE, Griffith JP, et al. (1994) Structure and mechanism of interleukin-1 beta converting enzyme. Nature 370: 270-275.
- Gu Y, Kuida K, Tsutsui H, Ku G, Hsiao K, et al. (1997) Activation of interferon-gamma inducing factor mediated by interleukin-1beta converting enzyme. Science 275: 206-209.
- Sagan L (1967) On the origin of mitosing cells J Theor Biol 14: 255-274.
- Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, Zorov DB (1988) Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J Cell Biol 107: 481-495.
- Liu R, Jin P, LiqunYu, Wang Y, Han L, et al. (2014) Impaired mitochondrial dynamics and bioenergetics in diabetic skeletal muscle. PLoS One 9: e92810.
- Elstner M, Andreoli C, Klopstock T, Meitinger T, Prokisch H (2009) The mitochondrial proteome database: MitoP2. Methods Enzymol 457: 3-20.
- Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, et al. (2003) Characterization of the human heart mitochondrial proteome. Nat Biotechnol 21: 281-286.
- DiMauro S1, Schon EA (2003) Mitochondrial respiratory-chain diseases. N Engl J Med 348: 2656-2668.
- Capaldi RA, Aggeler R (2002) Mechanism of the F(1)F(0)-type ATP synthase, a biological rotary motor. Trends Biochem Sci 27: 154-160.
- Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, et al. (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763-1766.
- Olichon A, Baricault L, Gas N, Guillou E, Valette A, et al. (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278: 7743-7746.
- Yoon Y, McNiven MA (2001) Mitochondrial division: New partners in membrane pinching. Curr Biol 11: R67-70.
- Gandre-Babbe S, van der Bliek AM (2008) The novel tail-anchored membrane protein Mff controls mitochondrial and peroxisomal fission in mammalian cells. Mol Biol Cell 19: 2402-2412.
- Hoppins S, Lackner L, Nunnari J (2007) The machines that divide and fuse mitochondria. Annu Rev Biochem 76: 751-780.
- Gilkerson RW, Margineantu DH, Capaldi RA, Selker JM (2000) Mitochondrial DNA depletion causes morphological changes in the mitochondrial reticulum of cultured human cells. FEBS Lett 474: 1-4.
- Legros F, Lombès A, Frachon P, Rojo M (2002) Mitochondrial fusion in human cells is efficient, requires the inner membrane potential, and is mediated by mitofusins. Mol Biol Cell 13: 4343-4354.
- Griparic L, Kanazawa T, van der Bliek AM (2007) Regulation of the mitochondrial dynamin-like protein Opa1 by proteolytic cleavage. J Cell Biol 178: 757-764.
- Guillery O, Malka F, Landes T, Guillou E, Blackstone C, et al. (2008) Metalloprotease-mediated OPA1 processing is modulated by the mitochondrial membrane potential. Biol Cell 100: 315-325.
- Ehses S, Raschke I, Mancuso G, Bernacchia A, Geimer S, et al. (2009) Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J Cell Biol 187: 1023-1036.
- Head B, Griparic L, Amiri M, Gandre-Babbe S, van der Bliek AM (2009) Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol 187: 959-966.
- Parone PA, Da Cruz S, Tondera D, Mattenberger Y, James DI, et al. (2008) Preventing mitochondrial fission impairs mitochondrial function and leads to loss of mitochondrial DNA. PLoS One 3: e3257.
- Chen H, Chomyn A, Chan DC (2005) Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem 280: 26185-26192.
- Nunnari J, Suomalainen A (2012) Mitochondria: in sickness and in health. Cell 148: 1145-1159.
- Tanner EA, Blute TA, Brachmann CB, McCall K (2011) Bcl-2 proteins and autophagy regulate mitochondrial dynamics during programmed cell death in the Drosophila ovary. Development 138: 327-338.
- Misawa T, Takahama M, Kozaki T, Lee H, Zou J, et al. (2013) Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nat Immunol 14: 454-460.
- Martinon F (2010) Signaling by ROS drives inflammasome activation. Eur J Immunol 40: 616-619.
- Zhou R, Yazdi AS, Menu P, Tschopp J (2011) A role for mitochondria in NLRP3 inflammasome activation. Nature 469: 221-225.
- Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, et al. (2012) Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36: 401-414.
- Furda AM, Marrangoni AM, Lokshin A, Van Houten B (2012) Oxidants and not alkylating agents induce rapid mtDNA loss and mitochondrial dysfunction. DNA Repair (Amst) 11: 684-692.
- Shokolenko I, Venediktova N, Bochkareva A, Wilson GL, Alexeyev MF (2009) Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res 37: 2539-2548.
- Bogenhagen DF, Rousseau D, Burke S (2008) The layered structure of human mitochondrial DNA nucleoids. J Biol Chem 283: 3665-3675.
- Kaufman BA, Durisic N, Mativetsky JM, Costantino S, Hancock MA, et al. (2007) The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol Biol Cell 18: 3225-3236.
- Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, et al. (2013) Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity 39: 311-323.
- Chu CT, Bayir H, Kagan VE (2014) LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons: implications for Parkinson disease. Autophagy 10: 376-378.
- Park S, Juliana C, Hong S, Datta P, Hwang I, et al. (2013) The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol 191: 4358-4366.
- Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN (2013) The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell 153: 348-361.
- Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y (2013) Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A 110: 17963-17968.
- Lee HK, Song JH, Shin CS, Park DJ, Park KS, et al. (1998) Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res Clin Pract 42: 161-167.
- Ritov VB, Menshikova EV, Azuma K, Wood R, Toledo FG, et al. (2010) Deficiency of electron transport chain in human skeletal muscle mitochondria in type 2 diabetes mellitus and obesity. Am J Physiol Endocrinol Metab. 298: E49-58.
- Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, et al. (2003) PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 34: 267-273.
- Patti ME, Butte AJ, Crunkhorn S, Cusi K, Berria R, et al. (2003) Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc Natl Acad Sci U S A 100: 8466-8471.
- Kadowaki T, Kadowaki H, Mori Y, Tobe K, Sakuta R, et al. (1994) A subtype of diabetes mellitus associated with a mutation of mitochondrial DNA. N Engl J Med 330: 962-968.
- Lindroos MM, Borra R, Mononen N, Lehtimäki T, Virtanen KA, et al. (2011) Mitochondrial diabetes is associated with insulin resistance in subcutaneous adipose tissue but not with increased liver fat content. J Inherit Metab Dis 34: 1205-1212.
- Bannwarth S, Procaccio V, Lebre AS, Jardel C, Chaussenot A, et al. (2013) Prevalence of rare mitochondrial DNA mutations in mitochondrial disorders. J Med Genet. 50:704-714.
- Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, et al. (2008) Prevalence of mitochondrial DNA disease in adults. Ann Neurol 63: 35-39.
- Feinstein R, Kanety H, Papa MZ, Lunenfeld B, Karasik A (1993) Tumor necrosis factor-alpha suppresses insulin-induced tyrosine phosphorylation of insulin receptor and its substrates. J Biol Chem 268: 26055-26058.
- Hotamisligil GS, Shargill NS, Spiegelman BM (1993) Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259: 87-91.
- Lee YS, Li P, Huh JY, Hwang IJ, Lu M, et al. (2011) Inflammation is necessary for long-term but not short-term high-fat diet-induced insulin resistance. Diabetes 60: 2474-2483.
- Shoelson SE, Lee J, Goldfine AB (2006) Inflammation and insulin resistance. J Clin Invest 116: 1793-1801.
- Henriksbo BD Lau TC, Cavallari JF, Denou E, Chi W1, et al. (2014) Fluvastatin causes NLRP3 inflammasome-mediated adipose insulin resistance. Diabetes.
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, et al. (2011) The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med 17: 179-188.
- Mirza RE1, Fang MM, Weinheimer-Haus EM, Ennis WJ, Koh TJ (2014) Sustained inflammasome activity in macrophages impairs wound healing in type 2 diabetic humans and mice. Diabetes 63: 1103-1114.
- Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, et al. (2003) Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac Myocytes. Circulation 107: 1418-1423.
- Samavati L, Lee I, Mathes I, Lottspeich F, Huttemann M (2008) Tumor necrosis factor alpha inhibits oxidative phosphorylation through tyrosine phosphorylation at subunit I of cytochrome c oxidase. J Biol Chem. 283: 21134-21144.
- Vadrot N, Ghanem S, Braut F, Gavrilescu L, Pilard N, et al. (2012) Mitochondrial DNA maintenance is regulated in human hepatoma cells by glycogen synthase kinase 3β and p53 in response to tumor necrosis factor a. PLoS One 7: 40879.
- Suliman HB, Welty-Wolf KE, Carraway MS, Schwartz DA, Hollingsworth JW, et al. (2005) Toll-like receptor 4 mediates mitochondrial DNA damage and biogenic responses after heat-inactivated E. coli. FASEB J 19: 1531-1533.
- Remels AH1, Gosker HR, Schrauwen P, Hommelberg PP, Sliwinski P, et al. (2010) TNF-alpha impairs regulation of muscle oxidative phenotype: implications for cachexia? FASEB J 24: 5052-5062.
- Wang M, Wang XC, Zhang ZY, Mou B, Hu RM (2010) Impaired mitochondrial oxidative phosphorylation in multiple insulin-sensitive tissues of humans with type 2 diabetes mellitus. J Int Med Res 38:769-781.
- Yuzefovych LV, Musiyenko SI, Wilson GL, Rachek LI (2013) Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS One 8: 54059.
- Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, et al. (2007) Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 131: 476-491.
- Park SY, Lee W (2007) The depletion of cellular mitochondrial DNA causes insulin resistance through the alteration of insulin receptor substrate-1 in rat myocytes. Diabetes Res Clin Pract 77 Suppl 1: S165-171.
- Ryu HS, Park SY, Ma D, Zhang J, Lee W (2011) The induction of microRNA targeting IRS-1 is involved in the development of insulin resistance under conditions of mitochondrial dysfunction in hepatocytes. PLoS One 6: e17343.
- Amuthan G, Biswas G, Zhang SY, Klein-Szanto A, Vijayasarathy C, et al. (2001) Mitochondria-to-nucleus stress signaling induces phenotypic changes, tumor progression and cell invasion. EMBO J 20: 1910-1920.
- Owusu-Ansah E, Yavari A, Mandal S, Banerjee U (2008) Distinct mitochondrial retrograde signals control the G1-S cell cycle checkpoint. Nat Genet 40: 356-361.
- Garbett K, Ebert PJ, Mitchell A, Lintas C, Manzi B, et al. (2008) Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol Dis 30: 303-311.
- Ziats MN, Rennert OM (2011) Expression profiling of autism candidate genes during human brain development implicates central immune signaling pathways. PLoS One. 6:e24691.
- Wei H1, Zou H, Sheikh AM, Malik M, Dobkin C, et al. (2011) IL-6 is increased in the cerebellum of autistic brain and alters neural cell adhesion, migration and synaptic formation. J Neuroinflammation 8: 52.
- Ashwood P, Wakefield AJ (2006) Immune activation of peripheral blood and mucosal CD3+ lymphocyte cytokine profiles in children with autism and gastrointestinal symptoms. J Neuroimmunol 173: 126-134.
- Molloy CA, Morrow AL, Meinzen-Derr J, Schleifer K, Dienger K, et al. (2006) Elevated cytokine levels in children with autism spectrum disorder. J Neuroimmunol 172: 198-205.
- Franchi L, Eigenbrod T, Núñez G (2009) Cutting edge: TNF-alpha mediates sensitization to ATP and silica via the NLRP3 inflammasome in the absence of microbial stimulation. J Immunol 183: 792-796.
- Tang G, Gutierrez Rios P, Kuo SH, Akman HO, Rosoklija G, et al. (2013) Mitochondrial abnormalities in temporal lobe of autistic brain. Neurobiol Dis 54: 349-361.
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, et al. (2013) NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature 493: 674-678.
- Bracey NA, Beck PL, Muruve DA, Hirota SA, Guo J, et al. (2013) The Nlrp3 inflammasome promotes myocardial dysfunction in structural cardiomyopathy through interleukin-1beta. Exp Physiol. 98:462-472.
- Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, et al. (2012) Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimers Dis. 30:617-627.
- Chaung WW, Wu R, Ji Y, Dong W, Wang P (2012) Mitochondrial transcription factor A is a proinflammatory mediator in hemorrhagic shock. Int J Mol Med 30: 199-203.
- Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, et al. (2013) Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg 258: 591-596.
- Sun S, Sursal T, Adibnia Y, Zhao C, Zheng Y, et al. (2013) Mitochondrial DAMPs increase endothelial permeability through neutrophil dependent and independent pathways. PLoS One. 8: 59989.
Citation: Gilkerson R, Materon L (2014) Two Roads Converging: Mitochondria and Inflammatory Signaling. J Clin Immunol Immunother 1: 004.
Copyright: © 2014 Luis Materon, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.