Understanding Mesangial Damage and Repair: Insights from an Experimental Model of Immunoglobulin Light Chain-Associated Mesangiopathy
*Corresponding Author(s):
Guillermo A HerreraDepartment Of Pathology, Cellular Biology And Anatomy And Obstetrics And Gynecology, University In New Orleans, Louisiana, United States
Tel:+3186755878,
Fax:+3186754541
Email:gherr1@lsuhsc.edu
Abstract
Mesangial injury represents a crucial event in the pathogenesis of light chain-associated glomerulopathies in patients with plasma cell dyscrasias. The glomerulopathic light chains interact with mesangial cells where purported receptors regulate the downstream cellular mechanisms that will be activated and result in glomerular alterations. The physicochemical and conformational characteristics of the abnormal light chains are primarily responsible for the downstream events affecting the mesangial milieu.
Different light chains are responsible for two diseases with diametrically opposite mesangial alterations: Light chain deposition disease which results in the expansion of the mesangium due to accumulation of matrix proteins not present in the normal mesangium and AL (light chain-associated) amyloidosis where the native mesangial matrix is replaced by fibrils (amyloid). In both cases there is enhancement of mesangial cell apoptosis and the altered mesangium has a marked decrease of mesangial cells, most undergoing apoptosis.
The repair of the damaged mesangium is difficult due to the absence of enough mesangial cells that can participate in the process and also the damage can be so extensive that the intrinsic processes that are available for repair (i.e. recruitment of stem cells from bone marrow and precursor stem cells in renal niches) cannot effectively carry out the recovery. Introducing exogenous mesenchymal stem cells represents a novel therapeutic avenue that has been experimentally tested with promising results.
In-vitro, ex-vivo and in-vivo animal models have been created to study these disorders providing excellent platforms to elucidate pathogenesis and provide insightful information that can be translated to the practice of renal pathology, as the In-vitro and invivo platforms corroborate each other. The article addresses how mesangial injury occurs in LCDD and AL-amyloidosis as examples of generic mesangiopathies and summarizes existing data regarding the experimental use of exogenous mesenchymal stem cells in mesangial healing.
Keywords
GLOMERULOPATHIC LIGHT CHAINS PRODUCE MESANGIAL INJURY
Approximately 85% of the monoclonal proteins in patients with plasma cell dyscrasia are associated with renal disease. Involvement of the tubular interstitial compartment is more common (70% of all cases) than that of the glomerular (about 30%) or vascular compartments. In some patients there is involvement of more than one compartment, but the manifestations usually are centered in one of the renal compartments [1-5].
Investigations performed in the last 20 years have provided detailed insights into how these abnormal light chains interact with the mesangium to produce glomerular injury. A number of experimental platforms have been used in our laboratory to delineate the sequence of events that results in glomerular alterations which are centered in the mesangium. In all these, light chains purified from the urine of patients with renal biopsy-proven conditions have been used. The glomerulopathies associated with monoclonal light chains are Light Chain Deposition Disease (LCDD) and AL (light chainrelated) amyloidosis, each creating completely different alterations in the mesangium. The findings that have been elucidated have paved the way for the design of novel therapeutic interventions aimed at stopping, slowing down or avoiding renal damage in patients with plasma cell dyscrasias.
HOW LIGHT CHAINS INTERACT WITH THE GLOMERULUS
Normal light chains are small molecular weight proteins that are filtered through the peripheral capillary walls and are delivered to the proximal tubules where they are catabolized by endosomal activity in the apical portions of the tubules and their amino acids are then returned to the circulation. Normal and tubulopathic light chains do not interact with the mesangium thus mesangial homeostasis is maintained intact.
Glomerulopathic light chains exert their initial or most significant pathogenic effect at the level of the mesangium. Although interactions occur early in the development of these disorders with the peripheral capillary walls, there is no doubt that the two main diseases involved in glomerular alterations focus on the mesangium as the center for pathogenetic development and progression, eventually leading to glomerulosclerosis/glomerular obsolescence. These light chains are attracted to the mesangium because of certain physicochemically and conformational characteristics that make them interact with the mesangial cells [6].
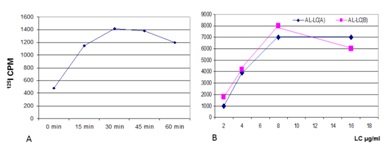
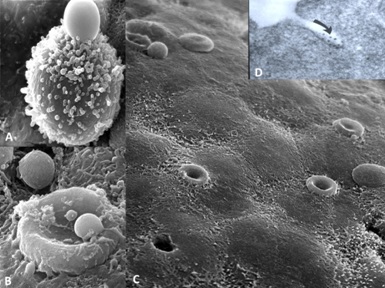
Studies have shown that there are saturable receptors on the mesangial cells that are utilized by the glomerulopathic light chains to effect their pathogenetic role (Figure 1) [7-8]. This receptor is not yet fully characterized and it appears to be of a high molecular weight (about 450-470 kd). It is not the cubilin-megalin receptor on proximal tubular cells used by light chains for their normal metabolism. When rat or human mesangial cells are incubated with glomerulopathic (but not tubulopathic) light chains, the number of caveolae on their surfaces increases dramatically [7]. The interactions of the glomerulopathic light chains occur with caveolae which are located on the mesangial cell surface (Figure 2). Caveolae play a role in signaling [8-11].
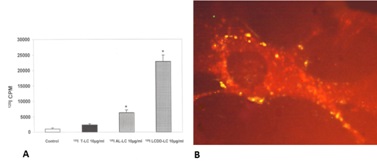
Both LCDD and amyloid forming light changes bind to receptors on mesangial cells and co-localize on the same sites along the membranes of mesangial cells, competing for the same receptor (Figure 3) [12]. However, while amyloidogenic light chains are avidly internalized into the mesangial cells (Figure 3B) using a clathrinmediated mechanism (Figure 4), this is not the case with LCDDlight chains which activate cellular mechanisms when they interact with the surface receptors but are only minimally internalized [13,14]. Interaction with the surface receptors is responsible for their pathogenic effects downstream (Figures 3-5).

INITIAL EFFECTOR MOLECULAR MECHANISMS THAT ARE ACTIVATED
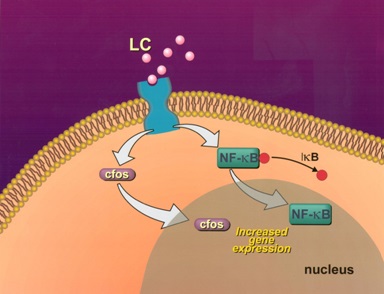
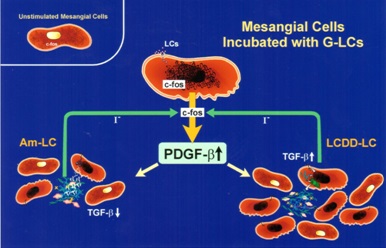
Interactions between glomerulopathic light chains and mesangial cells in culture (grown as monolayers on coverslips or on Matrigel) activate c-fos and NF-kβ which normally display a cytoplasmic localization in normal mesangial cells (Figure 5) and once stimulation occurs are translocated to the nuclei. C-fos has been found to participate in the activation of growth factors responsible for proliferative cellular activities (PDGF-β) and for cytoskeletal changes, including phenotypic transformation, in mesangial cells. NF-kβ plays a crucial role in regulating Monocyte Chemoattractant Protein (MCP)-1 production by mesangial cells [7].
GROWTH FACTORS PARTICIPATING IN THE DEVELOPMENT OF THESE RENAL DISEASES
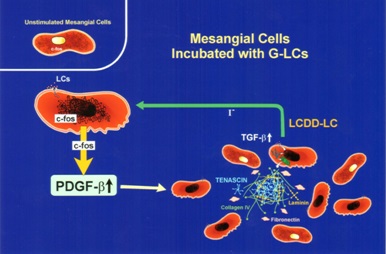
Initially, in both situations, there is production of PDGF-β by mesangial cells and activation leading to mesangial cell proliferation once c-fos signaling has occurred (Figure 6). Afterwards, the two conditions engage different growth factors in their progression as a result of the signaling mechanisms involved being different. In the case of LCDD, transforming growth factor (TGF)-β is activated and becomes actively secreted by the mesangial cells. This growth factor is fundamental in the production of extracellular matrix and also exerts an inhibitory effect on mesangial cell proliferation (Figure 7) [15-18]. In contrast, amyloid formation occurs when mesangial cells are incubated with amyloidogenic light chains, and amyloid potentiates metalloproteinases which destroy the native mesangium and INHIBIT TGF-β, preventing matrix rebuilding. The end result is abundant matrix deposition in LCDD and amyloid fibrils in the affected mesangium in AL-amyloidosis. The presence of these two fundamental growth factors has been documented in glomeruli of patients with the two entities, highlighting the translational value and repercussion of these studies [19].
HOW DIFFERENT GLOMERULOPATHIC LIGHT CHAIN AFFECT MESANGIAL CELL PHENOTYPE AND TRAFFIC IN MESANGIAL CELLS
When human or rat mesangial cells are incubated with glomerulopathic light chains extracted and purified from the urine of patient with renal biopsy-proven LCDD and AL-amyloidosis, there are dramatic changes that can be noted in each of the two conditions. These alterations in the mesangial "milieu" are crucial to understand the pathogenesis of these disorders and to translate the findings to clinical practice.
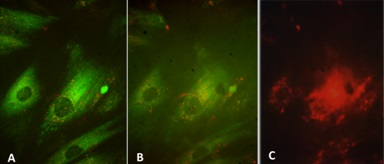
Normal mesangial cells exhibit characteristics of smooth muscle cells with abundant intracytoplasmic filaments maintained together by dense bodies or spindle densities (so-called myofilaments) and attachments plaques at their surface (Figure 8A,B). Normal mesangial cells express smoothelin and in some instances smooth muscle actin, the latter most noticeably when an activated phenotype develops. LCDD-light chains induce transformation of mesangial cells from their smooth muscle to a myofibroblastics phenotype (some consider this phenotype as an embryonic phenotype). This occurs by loss of some of the contractile elements and acquisition of cisternae of rough endoplasmic reticulum endowing the mesangial cells with the ability to increase their production of extracellular matrix proteins [16,20]. These activated mesangial cells typically express smooth muscle actin and maintain expression of smoothelin (Figure 8C).
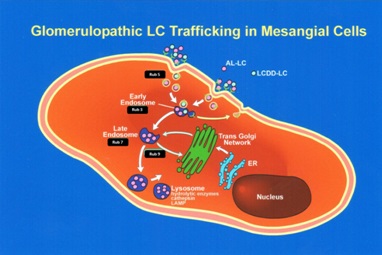
In contrast, the mesangial cells transform to a macrophage phenotype when incubated with amyloidogenic light chains (Figure 9). This change is morphologically characterized by the loss of contractile elements in their cytoplasm and the appearance of numerous lysosomes resulting in marked expansion of the lysosomal compartment by endowing these transformed mesangial cells with the machinery needed for active endocytosis and protein catabolism to take place. Rab proteins play a key role in transporting the light chains to the mature lysosomal compartment (Figure 10A) where they are processed and transformed into fibrils [12]. The transformed mesangial cells initially displayed Early Endosomal Antigen (EEA)- 1 and later Lysosomal - Associated Membrane Protein (LAMP) and cathepsin D (characteristic of mature lysosomes) in their cytoplasm (Figure 10B). These mesangial cells acquire phagocytic/macrophage markers such as CD68 and Lysotracker (Figure 11), as a result of the acquisition of numerous lysosomes. During the process of mesangial cell transformation, hybrid cells with macrophage / smooth muscle characteristics can be identified attesting to the in-situ cellular transformation process and mitigating against the idea that these macrophage like cells are transported from the bone marrow, as was suggested could happen in-vivo (Figure 12). This plasticity of mesangial cells is crucial to drive pathologic events that occur in association with monoclonal light chain-associated mesangiopathies [20].
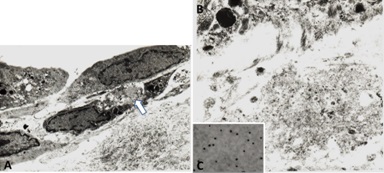
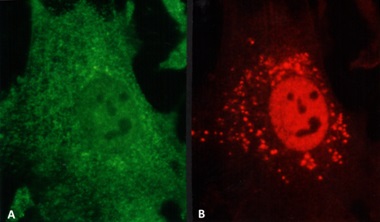
The two different types of glomerulopathic light chains though they use and compete for the same receptor on mesangial cells, exhibit entirely different intracellular trafficking patterns [12]. In the case of LCDD light chains, the minimal internalization that occurs leads to endosomal catabolism, with the receptor being returned to the cell membranes (Figure 13). The amyloidogenic light chains are taken in by the mesangial cells and the endosomal system is unable to catabolize them, directing the internalized light chains to the mature lysosomal system where catabolism leads to the formation of fibrils (Figures 11,13). The lysosomes must maintain an acidic environment to process the internalized light chains and convert them into fibrils. If the pH of the lysosomes is increased, their ability to form fibrils is impaired [21]. These fibrils are then extruded into the extracellular compartment when the lysosomes fuse with the cellular membranes and actively expel their contents [22].
WHAT ARE THE MORPHOLOGIC PARAMETERS THAT ALLOW TO MAKE A DIAGNOSIS OF THESE DISEASES IN THE CLINICAL ARENA?
It is important to understand how these diseases manifest their pathologic effects in the glomerular compartment to relate findings to those observed in experimental platforms. Renal biopsies from patients with LCDD and AL-amyloidosis reveal completely divergent mesangial abnormalities. Both diseases evolve from minimal manifestations to advanced disease and the sequence of events that must take place have now been elucidated in the laboratory where these diseases have been recreated for the purposes of studying the molecular mechanisms at play.
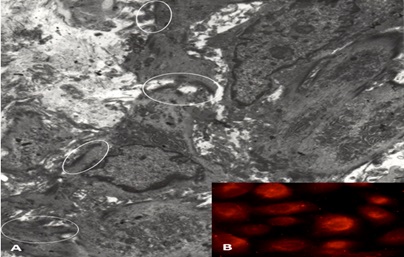
Patients with LCDD and renal involvement can present with a variety of glomerular manifestations depending on the stage of the disease process. This is easy to understand now that we have a clear appreciation of the events that must take place to arrive to the advanced morphologic expression known as nodular glomerulosclerosis (Figure 14), a process that mimics morphologically what is seen in advanced diabetic nephropathy, suggesting similar molecular mechanisms involved in their pathogenesis [18]. LCDD is preferentially associated with kappa light chains, mostly kappa I and IV, but there are cases also associated with lambda light chains [23-25]. These light chains manifest morphologically as punctate electron dense material that is sometimes (if not electron dense enough) difficult to identify, as it tends to blend with the increased mesangial matrix (Figure 14). While in the initial phases of LCDD there are proliferative mesangial changes mediated by PDGF-β, as the disease progresses and matures, mesangial matrix deposition dominates the pathologic picture, driven by TGF-β, which in turn exerts a negative feedback on mesangial cellular proliferative activity resulting in the formation of mesangial nodules and progressive loss of mesangial cells as a direct effect of the glomerulopathic light chains [14,17]. Depending on which growth factor predominates in the mesangial milieu at the time of the renal biopsy, the morphologic findings will vary accordingly with either more or less mesangial cell proliferation and/or matrix deposition [19].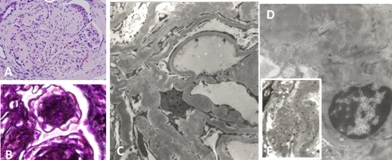
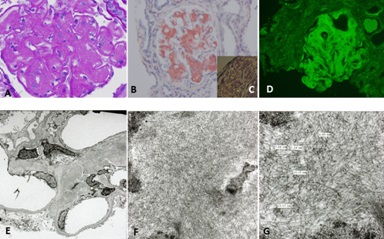
Patients with glomerular AL-amyloidosis progressively replace their normal mesangial matrix by non-branching fibrils with characteristic disposition (randomly disposed) and measuring 7-12 nm in diameter (Figure 15). This process; however, occurs slowly and in the early phases of this disease the replacement of normal mesangium may be very focal and segmental. The increased mesangial deposition of amyloid serves to further enhance apoptosis of mesangial cells which also occurs as a direct consequence of the amyloidogenic light chains [26-30]. Amyloidosis is characteristically associated with lambda light chains and glomerular amyloid is generally derived from ?6 light chains [31,32]. However, kappaassociated amyloidosis also occurs, though much less commonly.
EXTRACELLULAR MATRIX IN THESE DISORDERS
The experimental platforms clearly showed how the mesangial matrix is affected in these two conditions and the molecular mechanisms involved in the pathogenesis of these alterations. In LCDD, new matrix was produced by mesangial cells resulting in expansion of the mesangium. However, the matrix was noted to be abnormal. The normal collagen IV-rich mesangium was replaced by a tenascin-rich matrix. Genes normally quiescent in normal mesangial cells were activated leading to the production of abnormal extracellular matrix with fibrillary collagens, and most importantly, tenascin. Tenascin is a tenacious protein with an octopus-like appearance which binds strongly to integrins and matrix constituents [14]. It is a very difficult matrix protein to destroy and dispose of.
In the case of amyloidosis, it has been conclusively shown experimentally, that amyloid activate matrix metalloproteinases leading to destruction of the normal mesangial matrix which is then replaced by amyloid fibrils [30,33,34].
In both circumstances, mesangial cells become apoptotic (Figure 16) as a direct consequence of the monotypical glomerulopathic light chains, resulting in significant decrease and eventually almost total disappearance of mesangial cells [35]. This is clearly evident in experimental platforms and in renal samples from patients with advanced LCDD and AL-amyloidosis (Figures 14,15).
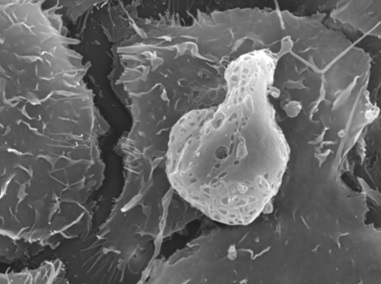
Figure 16: Apoptotic mesangial cell in mesangial cell culture incubated with amyloidogenic light chain (10µg/ml). Scanning electron microscopy X9800
REPRODUCTION OF PATHOLOGIC CHANGES IN THE MESANGIUM IN IN-VITRO, EX-VIVO AND IN-VIVO EXPERIMENTAL PLATFORMS
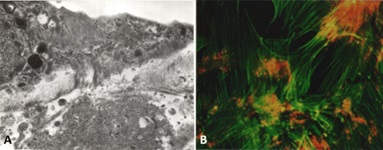
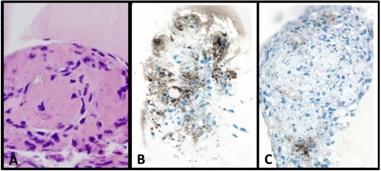
The various platforms allowed for complete reproduction of the two disease processes [14,15,30,34-37]. Mesangial nodularity with increased extracellular matrix proteins, predominantly tenascin (Figure 17) and replacement of the normal mesangial matrix by amyloid fibrils was observed in the In-vitro, ex-vivoand in-vivo models using LCDD and amyloidogenic light chains, respectively. Each one of these platforms permitted a unique appreciation of the events taken place. Using the 6D live image system, the sequence of events taking place could be clearly depicted and the process of repair could be evaluated sequentially to completion. Each of the models was particularly useful for various purposes. For example, the cell culture model using coverslips and Matrigel demonstrated the basic principles at play and were particularly useful to delineate early events (Figure 18) [37]. The ex-vivo model provided a well-controlled system to create mesangial damage [34,36] and then repair with monitoring of key elements, including amounts of glomerulopathic light chains and then mesenchymal stem cells delivered to the kidney [38]. Finally, the in-vivo animal model where samples were studied with transmission and scanning microscopy was unique in depicting the process of amyloid secretion into the extracellular matrix [22]. In conjunction, the models provided a unified conglomerate of data reproducing the key cellular events in each case and substantiated the fact that what was observed In-vitro also occurred in-vivo. This fact further demonstrated the translational nature of these studies [39] and the applicability to design therapeutic strategies for patients with glomerulopathies associated with plasma cell dyscrasias.
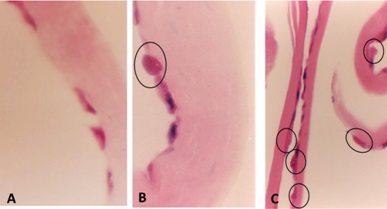
THE CHALLENGE OF REPAIRING THE DAMAGED MESANGIUM
If the glomeruli are not repaired, renal failure eventually occurs. As a result, the damaged mesangial areas in need of repair present several challenges: 1- Lack of mesangial cells to engage in the process; 2- Debris and abnormal material including fibrils and abnormal extracellular matrix proteins need to be disposed of, and 3-Lack of necessary growth factors, such as PDGF-β and TGF-β and other effector molecules to participate in and facilitate the healing process [38,40].
MECHANISMS OF MESANGIAL REPAIR
Once glomerular damage occurs, there are a number of intrinsic mechanisms that are activated which will try to heal the damage. These mechanisms include migration of bone marrow mesenchymal stem cells to the kidneys and/or activation of progenitor stem cells that are present in renal niches [41-43].
These intrinsic mechanisms are limited and may not be entirely effective when the damage surpasses a certain degree of severity. The end result then, if no other therapies are employed, is for the glomeruli to become sclerotic or obsolescent with accompanying loss of their function. Ultimately the patient develops irreversible renal failure.
EXOGENOUS MESENCHYMAL STEM CELLS IN THE REPAIR OF THE DAMAGED MESANGIUM
Obviously it is important to begin treatment early, before irreversible glomerular changes take place. Injection of exogenous mesenchymal stem cells will provide not only cells that can be engaged in the process of repair but also important paracrine effects that will make the repair process much more efficient [38, 44-46].
One challenge is to make sure that the exogenous mesenchymal cells are delivered in adequate amounts to the sites of injury. Interestingly, they do so quite effectively at least when perfused through the renal artery but it may be more challenging to accomplish the desired delivery to certain sites, if they are diluted in the circulation. Pre-administration of curcumin enhances the migration of mesenchymal stem cells to where they are needed for repair [39].
Exogenous stem cells migrate to sites of injury (Figure 19) and rapidly engage in cleaning the damaged mesangium by acquiring a macrophage phenotype and phagocytosing the extraneous material. Once cleaning has taken place, the mesenchymal stem cells which are typically primitive and poorly differentiated with minimal amounts of cytoplasm with few organelles, differentiate into mature mesangial cells acquiring more cytoplasm in the form of cell process, and contractile elements such as myofilaments and attachment plaques [39,45,46]. This sequence of events was clearly depicted using the 6D liver cell imaging system (Figure 20) and confirmed in the ex-vivo platform. They eventually are able to perform contractile functions [46] and are morphologically akin to normal mesangial cells. Intrinsic extracellular matrix properties regulate stem cell differentiation [47]. Then, transformed stem cells proceed to generate new mesangial matrix.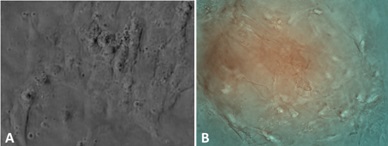
CONCLUSION
This manuscript summarizes how mesangial damage occurs in mesangiopathies and how it can be repaired, at least experimentally. The information that has been obtained should be of great value in the design of novel therapeutic avenues to address renal damage in plasma cell dyscrasias. The use of mesenchymal stem cells to repair the damaged mesangium appears to be a promising therapy to avoid irreversible renal damage in many glomerulopathies, not only those associated with plasma cell dyscrasias but also in other glomerular disorders with similar molecular pathogenesis, as the mechanisms involved are generic. Understanding mesangial damage represents a definitive advance in terms of future development of new therapeutic avenues to ameliorate or stop irreversible glomerulosclerosis.
REFERENCES
- Herrera GA (2000) Renal manifestations of plasma cell dyscrasias: an appraisal from the patients' bedside to the research laboratory.Ann DiagnPathol 4: 174-200.
- Markowitz GS (2004) Dysproteinemia and the kidney.AdvAnatPathol 11: 49-63.
- Sanders PW, Herrera GA, Kirk KA, Old CW, Galla JH (1991) The spectrum of glomerular and tubular interstitial lesions associated with monotypical immunoglobulin light chain deposition. Lab Invest 64: 527-537.
- Ronco PM, Aucouturier P (1999) The molecular bases of plasma cell dyscrasia-related renal diseases.Nephrol Dial Transplant 14: 4-8.
- Herrera GA (2009) Renal lesions associated with plasma cell dyscrasias: practical approach to diagnosis, new concepts, and challenges.Arch Pathol Lab Med 133: 249-267.
- Herrera GA (2003) Protein chemistry determines light chain-mediated renal damage in patients with plasma cell dyscrasias: a molecular understanding of variable manifestations. Chin J Pathol 32: 497-499.
- Russell WJ, Cardelli J, Harris E, Baier RJ, Herrera GA (2001) Monoclonal light chain-mesangial cells interactions: Early signaling events and subsequent pathologic effects. Lab Invest 81: 689-703.
- Keeling J, Herrera GA (2007) The Mesangium as a Target for Glomerulopathic Light and Heavy Chains: Pathogenic Considerations in Light and Heavy Chain-Mediated Glomerular Damage. In: Herrera GA (ed.). The kidney in plasma cell dyscrasias. Contributions to Nephrology series, S Karger Publishers, Basel, Switzerland. 153: 116-134.
- Cohen AW, Hnasko R, Schubert W, Lisanti MP (2004) Role of caveolae and caveolins in health and disease.Physiol Rev 84: 1341-1379.
- Anderson RG (1998) Thecaveolae membrane system.Annu Rev Biochem 67: 199-225.
- Thomsen P, Roepstorff K, Stahlhut M, van Deurs B (2002) Caveolae are highly immobile plasma membrane microdomains, which are not involved in constitutive endocytic trafficking.MolBiol Cell 13: 238-250.
- Teng J, Russell WJ, Gu X, Cardelli J, Jones ML, et al. (2004) Different types of glomerulopathic light chains interact with mesangial cells using a common receptor but exhibit different intracellular trafficking patterns.Lab Invest 84: 440-451.
- Keeling J, Teng J, Herrera GA (2004) AL-amyloidosis and light-chain deposition disease light chains induce divergent phenotypic transformations of human mesangial cells.Lab Invest 84: 1322-1338.
- Keeling J, Herrera GA (2009) An in vitro model of light chain deposition disease.Kidney Int 75: 634-645.
- Herrera GA, Russell WJ, Isaac J, Turbat-Herrera EA, Tagouri YM, et al. (1999) Glomerulopathic light chain-mesangial cell interactions modulate in vitro extracellular matrix remodeling and reproduce mesangiopathic findings documented in vivo.UltrastructPathol 23: 107-126.
- Teng J, Zhang PL, Russell WJ, Zheng LP, Jones ML, et al. (2003) Insights into mechanisms responsible for mesangial alterations associated with fibrogenicglomerulopathic light chains.Nephron Physiol 94: 28-38.
- Ronco P, Plaisier E, Mougenot B, Aucouturier P (2006) Immunoglobulin light (heavy)-chain deposition disease: from molecular medicine to pathophysiology-driven therapy.Clin J Am SocNephrol 1: 1342-1350.
- Zhu L, Herrera GA, Murphy-Ullrich JE, Huang ZQ, Sanders PW (1995) Pathogenesis of glomerulosclerosis in light chain deposition disease. Role for transforming growth factor-beta.Am J Pathol 147: 375-385.
- Herrera GA, Shultz JJ, Soong SJ, Sanders PW (1994) Growth factors in monoclonal light-chain--related renal diseases.Hum Pathol 25: 883-892.
- Herrera GA (2006) Plasticity of mesangial cells: a basis for understanding pathological alterations.UltrastructPathol 30: 471-479.
- Isaac J, Kerby JD, Russell WJ, Dempsey SC, Sanders PW, et al. (1998) In vitro modulation of AL-amyloid formation by human mesangial cells exposed to amyloidogenic light chains.Amyloid 5: 238-246.
- Teng J, Turbat-Herrera EA, Herrera GA (2014) Extrusion of amyloid fibrils to the extracellular space in experimental mesangial AL-amyloidosis: transmission and scanning electron microscopy studies and correlation with renal biopsy observations.UltrastructPathol 38: 104-115.
- Lin J, Markowitz GS, Valeri AM, Kambham N, Sherman WH, et al. (2001) Renal monoclonal immunoglobulin deposition disease: the disease spectrum.J Am SocNephrol 12: 1482-1492.
- Denoroy L, Déret S, Aucouturier P (1994) Overrepresentation of the V kappa IV subgroup in light chain deposition disease.ImmunolLett 42: 63-66.
- Picken MM, Frangione B, Barlogie B, Luna M, Gallo G (1989) Light chain deposition disease derived from the kappa I light chain subgroup. Biochemical characterization.Am J Pathol 134: 749-754.
- Herrera GA, Teng J, Turbat-Herrera EA (2011) Renal Amyloidosis: Current Views on Pathogenesis and Impact on Diagnosis. In: Herrera GA (ed.). Experimental Models for Renal Diseases: Pathogenesis and Diagnosis, S Karger Publishers, Basel, Switzerland. 169: 232-246.
- Keeling J, Dempsey S, Joseph L, Turbat-Herrera EA, Herrera GA (2005) The Role Of Matrix Metalloproteinases (MMPs) and Tissue Inhibitors of Metalloproteinases (TIMPs) in Mesangial Matrix Replacement in Al-Amyloidosis An in-vivo and In-vitro Correlative Study. In: Gilles Grateau, Robert AK, Martha Skinner (eds.). Amyloid and Amyloidosis. CRC Press, Boca Raton, Florida, USA. Pg: 136-138.
- vonGise H, Christ H, Bohle A (1981) Early glomerular lesions in amyloidosis. Electronmicroscopic findings.Virchows Arch A PatholAnatHistol 390: 259-272.
- Epstein WV, Tan M, Wood IS (1974) Formation of "amyloid" fibrils in vitro by action of human kidney lysosomal enzymes on Bence Jones proteins.J Lab Clin Med 84: 107-110.
- Keeling J, Herrera GA (2005) Matrix metalloproteinases and mesangial remodeling in light chain-related glomerular damage.Kidney Int 68: 1590-1603.
- Buxbaum JN, Chuba JV, Hellman GC, Solomon A, Gallo GR (1990) Monoclonal immunoglobulin deposition disease: light chain and light and heavy chain deposition diseases and their relation to light chain amyloidosis. Clinical features, immunopathology, and molecular analysis.Ann Intern Med 112: 455-464.
- Comenzo RL, Zhang Y, Martinez C, Osman K, Herrera GA (2001) The tropism of organ involvement in primary systemic amyloidosis: contributions of Ig V(L) germ line gene use and clonal plasma cell burden.Blood 98: 714-720.
- Keeling J, Herrera, GA (2008) Human matrix metalloproteinases: Characteristics and pathologic role in altering mesangial homeostasis. Microsc Res Tech 71: 371-379.
- Teng J, Herrera GA (2008) Mesangial amyloid formation in an isolated rat/kidney perfusion model: A new model to study renal amyloidogenesis. In: Lawreen HC, Martha S, John LB, David CS (eds.). XIth International Symposium on Amyloidosis. CRC Press, Boca Raton, Florida, USA.
- Tagouri Y, Sanders PW, Zhu L, Picken MM, Winokur TS, et al. (1995) Glomerulopathic light chains potentiate mesangial cell apoptosis In-vitro. Lab Invest 92: 161A.
- Herrera GA, Teng J, Liu X, Zhang Y, Turbat-Herrera EA (2004) Mesenchymal stem cells in mesangial repair in a model of immunoglobulin light chain-mediated mesangial injury. J Stem Cell Res Ther 4: 215.
- Tagouri YM, Sanders PW, Picken MM, Siegal GP, Kerby JD, et al. (1996) In vitro AL-amyloid formation by rat and human mesangial cells.Lab Invest 74: 290-302.
- Herrera GA, Teng J, Liu X, Zhang Y, Turbat-Herrera EA (2004) Mesenchymal stem cells in mesangial repair in a model of immunoglobulin light chain-mediated mesangial injury. J Stem Cell Res Ther 4: 215.
- Teng J, Turbat-Herrera EA, Herrera GA (2007) Role of translational research advancing the understanding of the pathogenesis of light chain-mediated glomerulopathies.PatholInt 57: 398-412.
- Herrera GA, Turbat-Herrera EA, Teng J (2011) Glomerular repair: Current status and future expectations. In: Herrera GA (ed.). Experimental models for renal diseases: Pathogenesis and Improving Diagnosis. Contributions to Nephrology series, S Karger Publishers, Basel, Switzerland. 169: 351-362.
- Rookmaaker MB, Smits AM, Tolboom H, Van 'tWout K, Martens AC, et al. (2003) Bone-marrow-derived cells contribute to glomerular endothelial repair in experimental glomerulonephritis.Am J Pathol 163: 553-562.
- Romagnani P (2011) Family portrait: renal progenitor of Bowman's capsule and its tubular brothers.Am J Pathol 178: 490-493.
- Nagy A, Quaggin SE (2010) Stem cell therapy for the kidney: a cautionary tale.J Am SocNephrol 21: 1070-1072.
- Kunter U, Rong S, Djuric Z, Boor P, Müller-Newen G, et al. (2006) Transplanted mesenchymal stem cells accelerate glomerular healing in experimental glomerulonephritis.J Am SocNephrol 17: 2202-2212.
- Wong CY, Cheong SK, Mok PL, Leong CF (2008) Differentiation of human mesenchymal stem cells into mesangial cells in post-glomerular injury murine model.Pathology 40: 52-57.
- Wong CY, Tan EL, Cheong SK (2014) In vitro differentiation of mesenchymal stem cells into mesangial cells when co-cultured with injured mesangial cells.Cell BiolInt 38: 497-501.
- Reilly GC, Engler AJ (2010) Intrinsic extracellular matrix properties regulate stem cell differentiation.J Biomech 43: 55-62.
Citation: Herrera GA, Turbat-Herrera EA, Teng J (2014) Understanding Mesangial Damage and Repair: Insights from an Experimental Model of Immunoglobulin Light Chain-Associated Mesangiopathy. J Cell Biol Cell Metab 1: 003.
Copyright: © 2014 Guillermo A Herrera, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.