Journal of Nephrology & Renal Therapy Category: Clinical
Type: Case Report
Uveitis and Late Onset Nephritis Associated With Henoch Schonlein Purpura: A Case Report
*Corresponding Author(s):
Sare Gülfem ÖZLÜFaculty Of Medicine, Department Of Pediatric Nephrology, Dr Sami Ulus Childrens And Maternity Hospital; Department Of Medicine, Yildirim Beyazit University, Yeni Ziraat Mahallesi, 657. Street 5/9, Alt?nda?, Ankara, Turkey
Tel:+90 5057735450,
Email:saredr@gmail.com
Received Date: Apr 22, 2016
Accepted Date: Jul 06, 2016
Published Date: Jul 20, 2016
Abstract
Henoch-Schönlein Purpura (HSP) is the commonest form of systemic small vessel vasculitis in children. It is characterized by non-thrombocytopenic purpura, arthritis and arthralgia, abdominal pain and gastrointestinal hemorrhage, and renal manifestations. Uveitis is rarely associated with HSP. To our knowledge there are only three patients in the adult literature and these patients had also renal manifestations of HSP. Recently the fourth case was reported in an 11-years-old boy, he had recurrent HSP but did not have renal manifestations. Here we report a pediatric case who had bilateral anterior uveitis and late onset severe renal involvement of HSP. We also tried to review the literature.
Keywords
Children; Henoch-schonlein purpura; Nephritis; Prognosis; Uveitis
INTRODUCTION
HSP is the most common systemic vasculitis of childhood and strongly associated with IgA deposition within small vessel walls. It usually involves skin, joints, gastrointestinal tract and kidneys. Etiology is unknown; genetic and environmental factors seem to play a role; and infection may be a potential trigger for the disease [1]. The characteristic vascular deposition of Immunoglobulin A (IgA) suggests that HSP is an IgA-mediated dysregulated immune response to antigen [1].
The disease usually occurs between the ages of 3 and 15 years and there is a slight male predominance. It is a self-limiting disease in the majority of children and prognosis is excellent [2]. Short-term morbidity and mortality is associated with gastrointestinal tract lesions and long-term prognosis is associated with severe renal involvement [2].
Uveitis can occur as manifestation of many rheumatological diseases most commonly Seronegative Spondyloarthritis (SSA), Behçet disease and juvenile idiopathic arthritis [3]. The association of HSP and uveitis is very rare and to our knowledge there are only four patients reported (Table 1). Three of these patients are in adult age and one is 11 years old [3-5]. Here we report an 8-years-old boy who presented with uveitis and late onset renal involvement of HSP and also reviewed the literature.
The disease usually occurs between the ages of 3 and 15 years and there is a slight male predominance. It is a self-limiting disease in the majority of children and prognosis is excellent [2]. Short-term morbidity and mortality is associated with gastrointestinal tract lesions and long-term prognosis is associated with severe renal involvement [2].
Uveitis can occur as manifestation of many rheumatological diseases most commonly Seronegative Spondyloarthritis (SSA), Behçet disease and juvenile idiopathic arthritis [3]. The association of HSP and uveitis is very rare and to our knowledge there are only four patients reported (Table 1). Three of these patients are in adult age and one is 11 years old [3-5]. Here we report an 8-years-old boy who presented with uveitis and late onset renal involvement of HSP and also reviewed the literature.
| Features | Kaur [3] | Erer [5] | Yamabe [4] | Muqit [6] | Our patient |
| Age at onset of uveitis | 11 years | 39 years | 64 years | 42 years | 13 years |
| Arthralgia | + | + | - | + | - |
| Rash | + | + | + | + | - |
| Uveitis | + | + | + | + | + |
| Recurrence of uveitis | + | + | - | - | - |
| Treatment | Prednisolone | Prednisolone and cyclophosphamide | Conservative | Prednisolone | Prednisolone and azathioprine |
CASE REPORT
An 8-year-old boy presented with rash, abdominal pain, vomiting and joint pain involving bilateral knees lasting for 24 hours. The child was previously healthy and he only had a history of upper respiratory tract infection 10 days ago. On physical examination he was 20kg (10th centile) in weight and 121cm (10thcentile) in height. His vitals including blood pressure were in normal limits. He did not have cyanosis, icterus, subcutaneous nodules, edema or lymphadenopathy. He had palpable purpuric rash involving markedly lower extremities. Bilateral knee joints were tender and swollen and the range of motion was decreased. He also had mild abdominal tenderness. Other systemic examinations were in normal limits. The child was diagnosed as Henoch Schonlein Purpura according to the EULAR criteria [7].
On laboratory investigation hemoglobin was 14.2g/dl, white blood cell count was 9900/mm3 platelet count was 417.000/mm3. C-Reactive Protein (CRP) was mildly elevated (15mg/dl, normal: 0-0,5); Erythrocyte Sedimentation Rate (ESR) was in normal limits. Renal function tests, urine microscopic analysis, liver function tests were normal. The Antinuclear Antibody (ANA) and rheumatoid factor were negative. Serology for Hepatitis B and C and Antineutrophil Cytoplasmic Antibody (ANCA) were negative. Throat swab was negative.
He was treated symptomatically by ibuprofen for arthralgia. Seven days later, the patient developed severe abdominal pain, nausea and vomiting. He had severe abdominal tenderness on physical examination. The laboratory tests showed elevated white blood cell count (18200/mm3) and CRP (29mg/dl) levels, and positive fecal occult blood test. There was no diagnostic finding on abdominal ultrasonography. Low dose steroids were given for a short duration for intestinal involvement. His clinical and laboratory findings improved gradually and he was discharged on the first week of hospitalization. Four weeks after the onset of the disease, asymptomatic mild proteinuria and microscopic hematuria were detected on urinalysis and continued for 8 weeks. Between 12 weeks -42 months during follow up, his physical examination and laboratory investigations were completely normal.
The patient was readmitted with pain and redness in his eyes 42 months after the onset of the disease. His physical examination was completely normal but ophthalmogical examination revealed bilateral anterior uveitis. There were no clinical signs and symptoms of renal involvement at this time. Fundus fluorescein angiography showed optic disc hyperfluorescence and capillary leakage. Visual acuity was intact in each eye. Differantial diagnosis for uveitis was investigated. There was no clinical and laboratory signs of infection and systemic inflammatory diseases. Acute phase reactants, serum complement levels, urinalysis, radiological examinations and echocardiography were all in normal limits. Antinuclear antibodies, anticytoplasmic antibodies and pathergy test were negative. Serum ACE levels were normal and he had no other sign and symptoms suggesting sarcoidosis. HSP was considered as a cause for uveitis, and azathioprine (2mg/kg/daily) and local steroids were started for treatment.
Six months later, he readmitted with rash and edema. Urinalysis showed hematuria and nephrotic range proteinuria. Hypoalbuminemia (3.3g/L) was detected. Glomerular filtration rate was 138ml/min/1.73m2and daily urinary protein loss 78mg/m2/h. ANA, anti ds-DNA antibodies, p- and c-ANCA were negative, and complement components normal. He was undergone renal biopsy because of heavy proteinuria and decrease in the serum albumin levels. Renal biopsy specimens showed diffuse mesengial proliferation and increase of the matrix (Figure 1), cellular (15/79 glomeruli) and fibrocellular crescents (1/79 glomeruli), global sclerosis (4/79 glomeruli), and fibrinoid necrosis and PMNL infiltration in a few glomeruli with deposition of mesangial IgA (+++/+++), C3 (++/+++) and fibrinogen (+/+++). These findings were defined as Grade III b HSP nephritis (diffuse mesangial proliferative glomerulonephritis) according to the classification of the International Study for Kidney Diseases in Children (ISKDC). We excluded tubulointerstitial nephritis and uveitis syndrome with renal biopsy findings. Ophthalmological examination revealed visual acuity exactly, anterior chamber scarring, peripheral vascular tortuosity in fundus (Clinical findings and treatment modalities are summarized in table 2). FFA was normal. He was administered oral prednisolone (2mg/kg/day) and ACE-?, and azathioprine was continued. At 3 months and 6 months follow up proteinuria improved and ocular findings were in normal limits.
On laboratory investigation hemoglobin was 14.2g/dl, white blood cell count was 9900/mm3 platelet count was 417.000/mm3. C-Reactive Protein (CRP) was mildly elevated (15mg/dl, normal: 0-0,5); Erythrocyte Sedimentation Rate (ESR) was in normal limits. Renal function tests, urine microscopic analysis, liver function tests were normal. The Antinuclear Antibody (ANA) and rheumatoid factor were negative. Serology for Hepatitis B and C and Antineutrophil Cytoplasmic Antibody (ANCA) were negative. Throat swab was negative.
He was treated symptomatically by ibuprofen for arthralgia. Seven days later, the patient developed severe abdominal pain, nausea and vomiting. He had severe abdominal tenderness on physical examination. The laboratory tests showed elevated white blood cell count (18200/mm3) and CRP (29mg/dl) levels, and positive fecal occult blood test. There was no diagnostic finding on abdominal ultrasonography. Low dose steroids were given for a short duration for intestinal involvement. His clinical and laboratory findings improved gradually and he was discharged on the first week of hospitalization. Four weeks after the onset of the disease, asymptomatic mild proteinuria and microscopic hematuria were detected on urinalysis and continued for 8 weeks. Between 12 weeks -42 months during follow up, his physical examination and laboratory investigations were completely normal.
The patient was readmitted with pain and redness in his eyes 42 months after the onset of the disease. His physical examination was completely normal but ophthalmogical examination revealed bilateral anterior uveitis. There were no clinical signs and symptoms of renal involvement at this time. Fundus fluorescein angiography showed optic disc hyperfluorescence and capillary leakage. Visual acuity was intact in each eye. Differantial diagnosis for uveitis was investigated. There was no clinical and laboratory signs of infection and systemic inflammatory diseases. Acute phase reactants, serum complement levels, urinalysis, radiological examinations and echocardiography were all in normal limits. Antinuclear antibodies, anticytoplasmic antibodies and pathergy test were negative. Serum ACE levels were normal and he had no other sign and symptoms suggesting sarcoidosis. HSP was considered as a cause for uveitis, and azathioprine (2mg/kg/daily) and local steroids were started for treatment.
Six months later, he readmitted with rash and edema. Urinalysis showed hematuria and nephrotic range proteinuria. Hypoalbuminemia (3.3g/L) was detected. Glomerular filtration rate was 138ml/min/1.73m2and daily urinary protein loss 78mg/m2/h. ANA, anti ds-DNA antibodies, p- and c-ANCA were negative, and complement components normal. He was undergone renal biopsy because of heavy proteinuria and decrease in the serum albumin levels. Renal biopsy specimens showed diffuse mesengial proliferation and increase of the matrix (Figure 1), cellular (15/79 glomeruli) and fibrocellular crescents (1/79 glomeruli), global sclerosis (4/79 glomeruli), and fibrinoid necrosis and PMNL infiltration in a few glomeruli with deposition of mesangial IgA (+++/+++), C3 (++/+++) and fibrinogen (+/+++). These findings were defined as Grade III b HSP nephritis (diffuse mesangial proliferative glomerulonephritis) according to the classification of the International Study for Kidney Diseases in Children (ISKDC). We excluded tubulointerstitial nephritis and uveitis syndrome with renal biopsy findings. Ophthalmological examination revealed visual acuity exactly, anterior chamber scarring, peripheral vascular tortuosity in fundus (Clinical findings and treatment modalities are summarized in table 2). FFA was normal. He was administered oral prednisolone (2mg/kg/day) and ACE-?, and azathioprine was continued. At 3 months and 6 months follow up proteinuria improved and ocular findings were in normal limits.
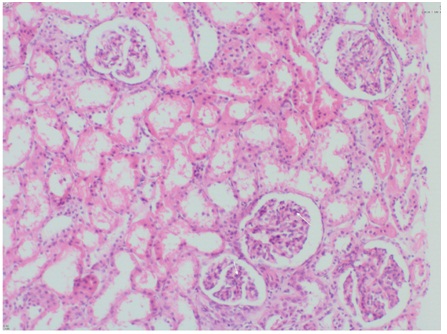
Figure 1: Renal biopsy specimen showing mesengial proliferation.
| Date | Organ involvement | Treatment |
| At admission | Only skin rash | Symptomatic |
| 1st week | Skin rash + gastrointestinal involvement | Oral steroids for one week(1mg/kg/daily) |
| 12th week | Normal physical examination | No treatment |
| 42nd week | Bilateral anterior uveitis | Azathioprine (2mg/kg/daily) |
| 48th week | Renal involvement (nephrotic syndrome) |
Azathioprine (2mg/kg/daily) |
Table 2: Treatment modalities according to organ involvement of our patient.
DISCUSSION
There are two important issues for this case that we have to emphasize on. First our patient had bilateral anterior uveitis associated with HSP. The association of HSP and anterior uveitis was first described by Yamabe et al., [4]. The pathogenesis of uveitis in HSP is tought to be related of the pathogenesis of the main disease [8]. There is alternative complement pathway activation in HSP and levels of abnormally glycosylated serum IgA1 are increased. These cannot be sufficiently cleared by liver and causes increased levels of IgA1 containing circulating immune complexes [1]. The immune complexes can reach the circulation of the eye and then they deposit in the uveal tissue. These complexes usually deposit on corneal endothelial cells, pigmented epithelial cells and vascular endothelial cells. Expression of adhesion molecules on these cells causes migration of leukocytes to the uveal tissue and cornea and results with tissue injury [7].
Although the patient reported in the study of Kaur et al., [3] did not have any renal involvement, all of the three patients reported in the adult population had varying degrees of renal involvement [4,5]. Our patient had minimal lesions such as hematuria on microscopic evaluation and 1+ proteinuria on dipstick examination on the fourth week of the disease. The other important issue we want to emphasize is the late onset renal involvement of our patient. He was completely well during four years of follow up; he developed uveitis in the third year of the disease and after six months of uveitis in the fourth year of disease proteinuria and microalbuminuria occurred. In general nephropathy develops within a few days to 4 weeks after onset of the symptoms. Findings include microscopic or gross hematuria, proteinuria, nephritic or nephrotic syndrome and occasionally progressive renal failure [9]. Retrospective studies detected that 20 to 54 percent of children with HSP had some degree of renal involvement [10]. Although most patients with HSP nephritis have mild and self limiting disease about 20% of children with HSP nephritis will develop chronic kidney disease, with up to 2% progressing end stage renal disease [1,10]. Narchi et al., [10] detected that renal manifestations occurred within four weeks in 84%, within six weeks in 91% and within six months in 97% of the patients and therefore they suggested that a six weeks of follow up will be sufficient. In a recent study of Tudorache E et al., [11] the authors found that even mild HSP nephritis had been associated with long term proteinuria. Ronkainen et al., [9] showed that 72% of patients in their cohort had renal symptoms after a mean follow-up of 19 years. In concordance with these studies our patient developed glomerulonephritis in the fourth year of the disease and in spite of immunosuppressive treatment.
In conclusion we would like to remind the importance of long term follow up in HSP even in the absence of significant renal findings at the beginning of the disease. Although it is not a part of routine examination in HSP; because of the possibility of uveitis we suggest an ophtalmological examination in these patients. Besides the paucity of the literature on this issue we speculate that uveitis may be more common in children with HSP nephritis.
Although the patient reported in the study of Kaur et al., [3] did not have any renal involvement, all of the three patients reported in the adult population had varying degrees of renal involvement [4,5]. Our patient had minimal lesions such as hematuria on microscopic evaluation and 1+ proteinuria on dipstick examination on the fourth week of the disease. The other important issue we want to emphasize is the late onset renal involvement of our patient. He was completely well during four years of follow up; he developed uveitis in the third year of the disease and after six months of uveitis in the fourth year of disease proteinuria and microalbuminuria occurred. In general nephropathy develops within a few days to 4 weeks after onset of the symptoms. Findings include microscopic or gross hematuria, proteinuria, nephritic or nephrotic syndrome and occasionally progressive renal failure [9]. Retrospective studies detected that 20 to 54 percent of children with HSP had some degree of renal involvement [10]. Although most patients with HSP nephritis have mild and self limiting disease about 20% of children with HSP nephritis will develop chronic kidney disease, with up to 2% progressing end stage renal disease [1,10]. Narchi et al., [10] detected that renal manifestations occurred within four weeks in 84%, within six weeks in 91% and within six months in 97% of the patients and therefore they suggested that a six weeks of follow up will be sufficient. In a recent study of Tudorache E et al., [11] the authors found that even mild HSP nephritis had been associated with long term proteinuria. Ronkainen et al., [9] showed that 72% of patients in their cohort had renal symptoms after a mean follow-up of 19 years. In concordance with these studies our patient developed glomerulonephritis in the fourth year of the disease and in spite of immunosuppressive treatment.
In conclusion we would like to remind the importance of long term follow up in HSP even in the absence of significant renal findings at the beginning of the disease. Although it is not a part of routine examination in HSP; because of the possibility of uveitis we suggest an ophtalmological examination in these patients. Besides the paucity of the literature on this issue we speculate that uveitis may be more common in children with HSP nephritis.
REFERENCES
- Brogan P, Bagga A (2011) Leukocytoclastic vasculitisin: Textbook of Pediatric Rheumatology, (6thedn), Saunders, Elsevier, Philadelphia. Pg: 483-497.
- Rieu P, Noël LH (1999) Henoch-Schönlein nephritis in children and adults. Morphological features and clinicopathological correlations. Ann Med Interne (Paris) 150: 151-159.
- Kaur S, Maheshwari A, Aneja S, Seth A, Beri S, et al. (2012) Henoch-Schönlein purpura with uveitis: an unusual case and review of literature. Rheumatol Int 32: 4057-4059.
- Yamabe H, Ozawa K, Fukushi K, Kubota H, Ohsawa H, et al. (1988) IgA nephropathy and Henoch-Schönlein purpura nephritis with anterior uveitis. Nephron 50: 368-370
- Erer B, Kamali S, Cingu K, Kilicaslan I, Gul A, et al. (2010) Recurrent anterior uveitis in Henoch Schonlein’s vasculitis. Rheumatol Int 30: 1377-1379
- Muqit MM, Gallagher MJ, Gavin M, Roberts F, Jardine AG (2005) Henoch-Schonlein purpura with keratitis and granulomatous anterior uveitis. Br J Ophthalmol 89: 1221-1222
- Ozen S, Pistorio A, Iusan SM, Bakkaloglu A, Herlin T, et al. (2010) EULAR/PRINTO/PRES criteria for Henoch-Schönlein purpura, childhood polyarteritis nodosa, childhood Wegener granulomatosis and childhood Takayasu arteritis: Ankara. Part II: Final classification criteria. Ann Rheum Dis 6: 798-806
- Chan CC, Li Q (1998) Immunopathology of uveitis. Br J Ophthalmol 82: 91-96
- Ronkainen J, Nuutinen M, Koskimies O (2002) The adult kidney 24 years after childhood Henoch-Schönlein purpura: a retrospective cohort study. Lancet 360: 666-670.
- Narchi H (2005) Risk of long term renal impairment and duration of follow up recommended for Henoch-Schonlein purpura with normal or minimal urinary findings: a systematic review. Arch Dis Child 90: 916-920
- Tudorache E, Azema C, Hogan J, Wannous H, et al. (2015) Even mild cases of paediatric Henoch-Schönlein purpura nephritis show significant long-term proteinuria. Acta Paediatr 104: 843-848.
Citation: ÖZLÜ SG, YAZILITA? F, AYDO? O, BÜLBÜL M, YILDIZ C, et al. (2016) Uveitis and Late Onset Nephritis Associated With Henoch Schonlein Purpura: A Case Report. J Nephrol Renal Ther 2: 007
Copyright: © 2016 Sare Gülfem ÖZLÜ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!