White Matter Hyperintensity and Vascular Disease from Biological Basis to Clinical Significance
*Corresponding Author(s):
Paolo GarofaloDepartment Of Radiology, Azienda Ospedaliero Universitaria, Di Cagliari - Polo Di Monserrato, S.S. 554 Monserrato (Cagliari) 09045, Italy
Email:paolo.garofalo89@gmail.com
Abstract
“White matter disease” identifies a series of different conditions and pathological mechanisms: autoimmune, infectious, toxic-metabolic and vascular. Each of these leads to a global impairment of the neural myelination process through the secondary destruction of previously myelinated structures. To date, the imaging spectrum represents an irreplaceable tool to detect these lesions, describe their distribution patterns and stage them over time. This study aims presents a pictorial review of white matter disease, from pathology to imaging spectrum, reporting current main staging systems with greater emphasis to relationship between cerebrovascular disease and white matter hyper-intensity appearance, and the newest advances in this field.
Keywords
CT = Computed Tomography; DWMH = Deep White Matter Hyperintensities; LA = Leukoaraiosis; MRI = Magnetic Resonance Imaging; PWMH = Periventricular White Matter Hyperintensities; SVD = Small Vessels Disease; WMH = White Matter Hyperintensities
INTRODUCTION
“White Matter Disease”, formerly described as “leukoaraiosis” by Hatchinsky on Computed Tomography (CT) [1], can be depicted into two main types according to their causes: a) primary, derived from an unknown etiology and b) secondary, derived from a great variety of known etiologies [2]. White Matter Hyperintensity (WMH) is a purely descriptive term currently used on Magnetic Resonance Imaging (MRI), and it represents a very common finding in older patients affected by a wide range of diseases included autoimmune, infective, toxic-metabolic and cerebrovascular. WMH can be classified into Periventricular (PV-WMH) and Deep Subcortical (DS-WMH). PV-WMH is characterized by gliosis, loosening of the white matter fibers and myelin, loss around tortuous venules in perivascular spaces, whereas the main features of DS-WMH are demyelination, gliosis and increased tissue loss as the lesions become more severe. Although cerebral blood flow and the cerebrovascular endothelial surface area are both so large as to consent storage of fluid and proteins within interstitial spaces, these findings suggest that Blood Brain Barrier (BBB) impairment plays a key role on genesis of WM damage [3].
Several papers demonstrated that specific clusters of human perivascular tissue proteins in the WM would be involved in the process of arteriolar wall thickening. One of these clusters includes fibrinogen, immunoglobulins and thrombomodulin, which occur in subcortical grey and white matter [4]; another cluster includes albumin, PCR (not defined), homocysteine, interleukin-6 and the intracellular adhesion molecule-1, diffusively present in cerebral interstitial tissue in patients with increased risk of WHM development and lacunar stroke [5]. Cerebrovascular diseases are related to BBB damage and to WMH through a series of concatenated events. From a pathological point of view, acute subcortical infarct leads, in 90% of cases, to cavitation or narrowing of surrounding brain tissues and secondary gliotic reaction that could create a protective “white matter hyperintensities cap” [6]. The main vascular pathologies associated with WMH are shown in table below (Table 1) [7].
|
|
Etiology |
Damaged Area |
|
Small Vessel Disease (SVD) |
Hereditary/Sporadic |
Basal ganglia; fronto-temporal and periventricular WM |
|
Amyloid-related angiopathy |
Hereditary |
Cortical-subcortical temporal lobe |
|
PrimaryAngitis of CNS |
Idiopathic |
Non-specific distribution |
|
CADASIL |
Hereditary |
Bilateral subcortical white matter of temporal lobe; bilateral external capsule |
|
CARASIL |
Hereditary |
Basal ganglia and thalamus; diffuse leukoencephalopathy; middle cerebellar peduncle across the pons (arch sign) |
|
MELAS |
Hereditary |
Parieto-occipital lobes with subcortical calcification |
|
SusacSyndrome |
Idiopathic |
Corpus Callosum |
Table 1: Main cerebrovascular disease causing WMH.
Small Vessels Disease (SVD), includes two main forms of cerebrovascular sicknesses: Arteriolosclerosis and Cerebral Amyloid Angiopathy. SVD due to Arteriolosclerosis relates to hypertension with muscle loss from the media with a general thickening and stiffness of the arterial wall and genesis of microaneurysm and micro-atheroma associated with calcification [8]. SVD due to Cerebral Amyloid Angiopathy is characterized by a progressive storage of the beta-amyloid protein into small and medium size cells of the vessel muscle wall mainly located on the leptomeningeal space, cortex and also in capillaries [9]. When vascular injuries occur in vascular “end-zones”, WMH distribution pattern will be focal beginning or diffuse confluent, while this pattern will appear centripetal in the basal nuclei, centrum semiovale, corona radiata and brainstem (Figure 1 and 2) [10].
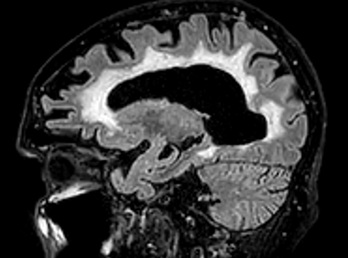 Figure 1: 77-yrs female with arteriolosclerosis-related SVD sagittal view on FLAIR-T2 sequences showing diffuse sovra-tentorial white matter hyperintensity.
Figure 1: 77-yrs female with arteriolosclerosis-related SVD sagittal view on FLAIR-T2 sequences showing diffuse sovra-tentorial white matter hyperintensity.
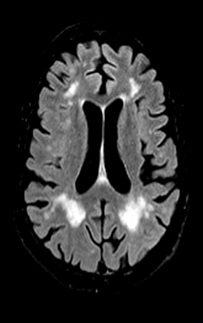 Figure 2: Axial view on T2-FLAIR of watershed zones infarcts showing multiple white matter hyperintensity areas along ACA-MCA (anteriorly) and MCA-PCA (posteriorly) border zones.
Figure 2: Axial view on T2-FLAIR of watershed zones infarcts showing multiple white matter hyperintensity areas along ACA-MCA (anteriorly) and MCA-PCA (posteriorly) border zones.
Further, the recent small subcortical infarcts (involving perforating arterioles) may evolves into rounded or ovoidal lacunes of 3-15 mm and represent the interrelate pathogenic substrate for WMH, which can be identified at the edge of ischemic territory [11]. CADASIL represents the most common hereditary cause of cerebral vascular pathology related to subcortical infarcts and leukoencephalopathy. WMH involves firstly, the temporal lobe; and then progress to the posterior temporal, frontal and parietal regions likely to the basal nuclei and thalamus [12]. PACNS is a very rare vasculitis syndrome, characterized by leptomeningeal and cortical vessels inflammation [13]. MRI shows different and nonspecific pattern: a) “hemorrhagic form” related to intra- parenchymal infarction; b) “pseudo-tumoral form” related to mass lesion with central necrosis, perilesional edema, infiltrating and surrounding cerebral structures. Hence angiography can demonstrate multifocal narrowing of large cerebral vessels [14].
Susac Syndrome is a rare and multifactorial disease that involves small vessels and leads to vascular occlusion with thrombus formation involving the thalamus, basal nuclei and subcortical white matter [15]. MELAS is hereditary syndrome due to mutations on the mitochondrial genome. MRI shows some signal alteration areas as hyperintense through T2 and FLAIR sequences frequently located on parieto-occipital cortex and less frequently in the temporo-parietal cortex. Characteristically, these lesions don’t respect the vascular distribution unlike ischemic lesions [16].
IMAGING HALLMARKS
CT imaging
Hatchinsky in the 1980 was the first scientist who described on CT, some findings of irregular low attenuation signals in the periventricular and deep white matter [1]. The different location of damage probably depends on different pathological pathways which involve different levels of brain vascularization. In particular, SVD determines Deep subcortical White Matter Hyperintensity (DWMH) due to the higher grade of hypoxic sensitivity along the so called “border zones,” with subsequent appearance of ischemic lacunas that are visible as hypo-intense areas on CT (Figure 3 and 4). On the contrary, especially on chronic stage, the involvement of large vessels causes a variable and sporadic distribution pattern of WM damage [17]. Periventricular White Matter Hyperintensity (PWMH) relates to expansion of pre-existing periventricular damage due essentially to benign processes such as venous collagenosis involving an elderly cohort of patients [18]. Otherwise, brain CT scan showed a high capability to localize white matter changes as one or more hypo-intensity areas, based on European Task Force on age related white matter changes, allowing staging of them into a four points scale [19].
0 = no lesion
1 = focal lesion > 5mm
2 = early confluent lesion
3 = diffuse involvement of brain
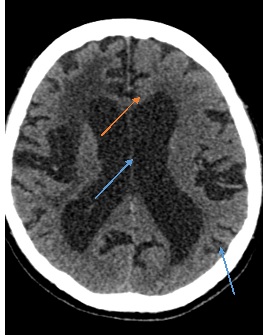 Figure 3: Multiple hypointensity(orange arrow) areas associated with enlarged cortical sulci (blue arrow) and ventricular spaces depicting hypoxic-ischemic encephalopathy.
Figure 3: Multiple hypointensity(orange arrow) areas associated with enlarged cortical sulci (blue arrow) and ventricular spaces depicting hypoxic-ischemic encephalopathy.
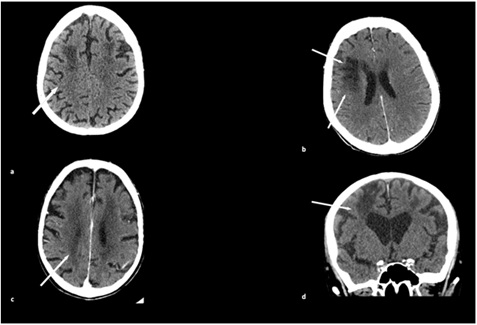 Figure 4: CT axial scans show large and confluent hypodensity «leukoaraiosis» areas related to moderate-severe SVD involving corona radiata and centrum semiovale with gliotic deterioration (white arrow).
Figure 4: CT axial scans show large and confluent hypodensity «leukoaraiosis» areas related to moderate-severe SVD involving corona radiata and centrum semiovale with gliotic deterioration (white arrow).
MRI imaging
WMH is most commonly detected using MRI sequences such as T2-FLAIR and Diffusion Tensor Imaging weighted (DTI) sequencesT2-weighted and T2- FLAIR sequences are able to show WMH. FLAIR sequence, in particular, using an attenuating CSF signal, can easily detect PWMH (Figure 1 and 2). Moreover, FLAIR can identify lacunar infarcts appearing as hypo-intense areas with a hyper-intense peripheral edge; these findings could represent a form of late-stage macroscopic damage. DTI sequences visualize orientation to quantify integrity of neuronal pathways of white matter with evaluation of Fractional Anisotropy (FA) and Mean Diffusivity (MD). FA is a one dimensional value scaled from 0 to 1 which describes the presence (FA=1) or not (FA=0) of unidirectional water diffusion. MD, in addiction with its two components Radial (RD) and Axial Diffusivity (AD), can respectively quantify parallel or perpendicular diffusivity processes to orientation of WM tracts [20]. Therefore, DTI can detect any microstructural damage of WM due to the demyelination process predating axonal loss [21].
Perfusion MR can dynamically evaluate WM lesions through BBB permeability measurement with two modalities:
• Arterial Spin Labeling: it works applying the inversion of longitudinal magnetization of blood flow of afferent artery without the use of contrast media. This inversion pulselabels specific intracranial vessels sampling in the head [22]
• Paramagnetic Contrast Administration = it works by reducing relaxation time on vessels and surrounding tissues that show an increase of signal on T1-weighted and a loss of signal on T2-T2*
Recent advances in MRI allow the more careful description of the location but also the nature (ischemic or not) of white matter injuries. Hydrogene Magnetic Resonance Spectroscopy (H-MRS) shows a reduced level of N-Acetyl Aspartate (NAA) and Cr along ischemic WMH area, which is intended as axonal and neuronal injury markers, while they do not show any variation in normal elderly patient’s cohort [23]. Another interesting role of MRI analyses is the evaluation of BBB integrity. Dynamic Contrast-Enhanced MRI (DCEMRI) can evaluate and stage compromised permeability of BBB alongside the ischemic core and “penumbra region” in deep white matter. DCEMRI identify increased permeability signal surrounding WMH’s area and decreased permeability signal within WMH’s area [24]. More recently, the 7 Tesla coils MRI yields a better spatial resolution to well identify early WM injuries, detecting more detailed sub cortical infarcts, micro bleeds and therefore distinguishing ischemic lacunas from perivascular spaces [25].
|
Imaging Technique |
Common Features |
|
Computed tomography |
Multiple and/or single hypo-dense lacunes involving periventricular and deep white matter |
|
Magnetic resonance |
Focal or diffuse areas of T2-FLAIR hyperintensity and T1 hypo-intensity; post-ischemic lacunes appear as a focal area of FLAIR hypo-intensity |
|
Diffusion tensor imaging |
Visualize orientation to quantify integrity of neuronal pathways of white matter with evaluation of Fractional Anisotropy (FA) and Mean Diffusivity (MD) |
|
Perfusion imaging |
Evaluate and stage compromised permeability of BBB alongside the ischemic core and “penumbra region” in deep white matter |
|
Spectroscopy |
Shows a reduced level of N-acetyl aspartate (NAA) and Cr along ischemic WMH area |
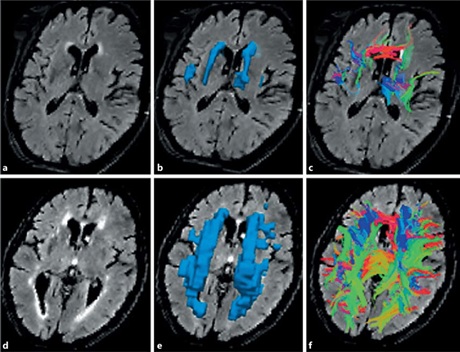
Quantitative tractography of fibers crossing WMH with small and large volumes of WMH.
Low-volume WMH (a-c) and high-volume WMH (d-f ). a, d T2 FLAIR MRI with WMH. b, e Three-dimensional region of interest of WMH. c, f Tracts crossing WMH. With kindly permission of Reginold, William & Luedke, Angela & Tam, Angela & Itorralba, Justine & Fernandez-Ruiz, Juan &Reginold, Jennifer & Islam, Omar & Garcia, Angeles. (2015).
MRI grading scales
Some Authors tried to define the location and size of white matter damage trying to relate this one to progression of underlying pathology, as Fazekas did in 1987, when he proposed the “Fazekas Scale” to simplify the quantification of amount of WMH lesions [26]. This scale is still valid, and it remains a very useful and direct system to classify the severity of white matter diseases according to size, location and confluence of lesions (Table 2). Later, in order to classify white matter changes, Wahlund (Table 3) and Longstreth (Table 4) assessed other two minor methods that included basal ganglia and both DWMH and PWMH involvement. [27,28]. The width of the subarachnoid spaces and lateral ventricles are classified with the use of analog visual rating scale.
|
Deep White Matter (DWMH) |
|||
|
P=0 |
P=1 |
P=2 |
P=3 |
|
Absent |
“caps” or pencil thin lining |
smoothhalo |
irregular periventricular signal extending into deep white matter |
|
Periventricular White Matter (PVWMH) |
|||
|
P=0 |
P=1 |
P=2 |
P=3 |
|
Absent |
punctate foci |
beginningconfluence |
large confluentareas |
Table 2 (Fazekas Scale): Four-point scale to describe deep white matter and periventricular white matter damage.
|
White Matter |
Number of Lesions |
|
0 |
Absent |
|
1 |
Focal |
|
2 |
Beginningconfluent |
|
3 |
Diffuse of entire regions with or without subcortical U-fibers |
|
BasalGanglia |
Number of Lesions |
|
0 |
Absent |
|
1 |
1 focallesion (>5 mm) |
|
2 |
More than 1 focallesions |
|
3 |
Confluentlesions |
Table 3 (Wahlund Scale): 4-point scale where focal lesions are not defined; they are interpreted as diffuse-focal lesion. Brain areas used for rating are: frontal, parieto-occipital, temporal, infratentorial and basal ganglia (globus pallidum, nucleus caudatus, putamen, thalamus, internal-external capsule and insula).
|
P=0 |
Absent |
|
P=1 |
Discontinuous periventricular rim with minimal dots of subcortical disease |
|
P=2 |
Thin continuous periventricular rim with a few patches of subcortical disease |
|
P=3 |
Thicker continuous periventricular rim with scattered patches of subcortical disease |
|
P=4 |
More irregular periventricular rim with mild subcortical disease and minimal confluent periventricular white matter hyperintensities |
|
P=5 |
Mild periventricular confluence surrounding the frontal and occipital hornes |
|
P=6 |
Moderate periventricular confluence surrounding the frontal and occipital hornes |
|
P=7 |
Periventricular confluence with moderate involvement of centrum semiovale |
|
P=8 |
Periventricular confluence involving most of the centrum semiovale |
Table 4 (Longstreth Scale): Eight-point scale for WM lesions graded at the level of the body of lateral ventricle.
Furthermore, Prins et al., [29], classified white matter lesions on the basis of their diffusion upon brain areas. This required defining periventricular a WMH area as directly extending along the ventricle’s borders and subcortical a WMH area as extending perpendicularly more or less 10 mm from the ventricle’s borders. This scale provides different scores depending on where the lesions are being located and their progression. According to this method, white matter lesions frame could include three main periventricular locations (frontal, occipital caps and lateral bands), and periventricular score fluctuates from -3 to +3; similarly, subcortical lesions includes four patterns of distribution (frontal, parietal, temporal and occipital) resulting in a subcortical score from -4 to +4.
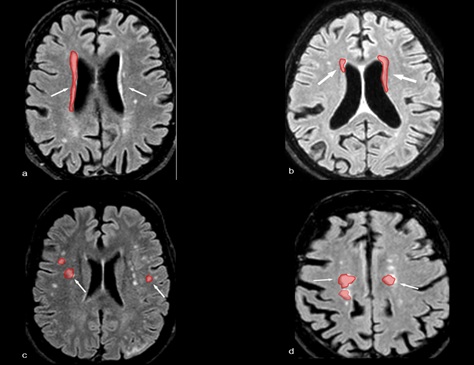
Schematic “mild” WMH distribution along periventricular and deep subcortical brain areas using different Staging Scales. Red shadows highlight scattered WMH areas.
Corresponding WMH Score:
|
Figure a |
Figure b |
Figure c |
Figure d |
|
Fazekas scale = 1 |
Fazekas scale = 2 |
Fazekas scale= 1 |
Fazekas scale = 2 |
|
Wahlund scale = 1.5 |
Wahlund scale = 1.5 |
Wahlund scale= 1.5 |
Wahlund scale = 2.5 |
|
Longstreght scale = 2 |
Longstreght scale = 1 |
Longstreght scale= 3 |
Longstreght scale = 4 |
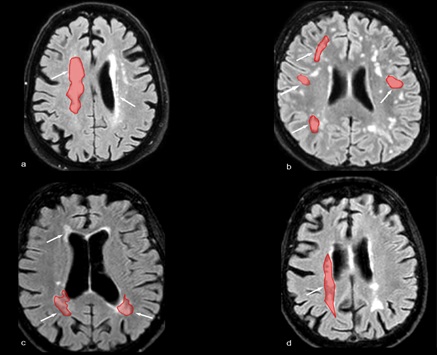
Schematic severe WMH distribution along periventricular and subcortical brain areas using different staging scales. Red shadows highlight confluent WMH areas.
Corresponding WMH Score:
|
Figure a |
Figure b |
Figure c |
Figure d |
|
Fazekas scale = 3 |
Fazekas scale = 3 |
Fazekas scale= 3 |
Fazekas scale = 3 |
|
Wahlund scale = 2.5 |
Wahlund scale = 2.5 |
Wahlund scale= 2 |
Wahlund scale = 2.5 |
|
Longstreght scale = 4 |
Longstreght scale = 4 |
Longstreght scale= 2 |
Longstreght scale = 4 |
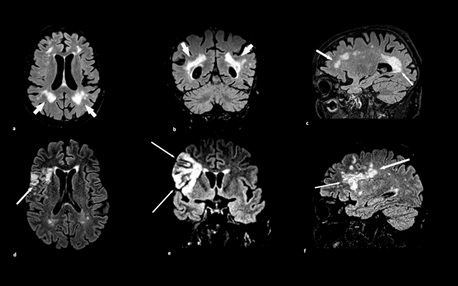
MRI different scanning plans obtained by FLAIR T2-weighted sequences showing large and confluent white matter hyperintensity areas related to advanced stages of SVD (Fazekas scale 3).
Figure a,d= axial scans show deep periventricular and subcortical partially confluent WMH areas extending to right frontal cortical sulci
Figure b,e = coronal scans show deep periventricular and subcortical largely confluent WMH extending to right parietal-temporal lobe
Figure c,f = sagittal scans show largely diffuse WMH extending from anterior to posterior involving splenium of corpus callosum and deep WM of frontal lobe
DISCUSSION AND CONCLUSION
WMH lesions tend to appear heterogeneous, reflecting their degree of “whiteness”. This feature explains how cellular breakdown leads to tissue structural and vascular changes not only inside of “core” of WMH area, but also on surrounding white matter’s “penumbra” which evolve further away from the visible area of WMH. In particular, the extension of WMH edges relates to the improvement of functional outcomes in patients with a previous acute ischemic stroke or cerebral hemorrhage [30]. Saba et al., investigated a probable intrinsic relationship between LA and the distribution of cerebral micro bleeds, intended as an independent biomarker for small vessel damage. Therefore they enrolled a cohort of 85 vasculopathic patients of unknown cause who submitted to MRI multiple GRE (gradient echo) T2* weighted scans and observed a statistically significant association between CMB’s, looking like small “dot-like” hypo intense lesions on GRE, and spreading WMH areas especially along periventricular brain regions [31].
According to these results, Lin et al., conducted a recent meta-analysis by enrolling more than 6912 participants with a past ischemic stroke treated with an rt-PA thrombolytic therapy in acute stage, trying to identify eventual relationships between the rate of extension of WMH burdens and clinical outcome after administration of the therapy [32]. They confirmed that deep micro bleeds detected by T2*-weighted gradient echo sequences relate to the periventricular hyper-intensity, suggesting a sensibility to post-recanalization blood flow necrosis in those areas in which there is an increased hyper-intensity signal. In particular, their results showed a double increased risk of intracerebral hemorrhage in acute ischemic stroke patients with detected LA. Therefore, the importance of detecting “leukoaraiosis” signs regards not only clinical outcome after stroke, but also the recovery after administration of rt-PA. Bahrani et al., proposed another interesting research perspective about WMH pathogenesis; they speculated about the specific relationship between WMH distribution (periventricular or deep) and decreased regional cortical blood flow. They found two important markers: the first was that deep WMH is associated with decreased regional cortical blood flow, and the second that blood flow in WM shows a greater reduction in the periventricular than in deep WMH areas. The strongest and most consistent risk factors that showed the clearest associations with WMH were age, hypertension, hypercholesterolemia, diabetes and smoke [33].
Despite the fact that many Authors considered hyperglycemia as having a primary role on genesis of SVD, there are many controversial associations between diabetes and WMH areas genesis, thus there is not sure concordances to consider diabetes as independent risk factor. This data discordance is likely due to a different and non- uniform sampling of case-control population [34]. DWMH or PVWMH can associate to different pathological correlates such as vascular dementia [35]. This particular form of dementia showed high relationships with subcortical impairment of the frontal white matter network and other single infarction areas with puncate or confluent “area foci”: thalamus, angular gyrus, deep frontal areas and the left hemisphere [36]. Even the small vessels circulation may be different between deep and periventricular areas, with higher periventricular white matter damage due to large vessels disease [37]. SVD leads to lacunar infarction that shows a tight connection to WHM. In particular, WMH proximal to normal course of perforating vessels, are highly distributed at the edge of incident lacunas and enlarge from periventricular to subcortical region [11]. These findings suggest that vascular risk factors with reduction of general cerebral blood flow are associated with a pervasive SVD involving both periventricular and deep white matter.
Recently, many Authors suggested a determinant role of carotid disease [38,39] as specific predictor factor for increasing WMH areas. In particular, Lucatelli et al., carried on a study considering a leading role of carotid occlusion degree, evaluating in particular carotid Intima-Media Thickness (IMT) and Intima-Media Thickness Variability (IMTV) as the driving parameters for cerebrovascular risk such as atherosclerosis progression or regression. They enrolled a cohort of 61 patients to MRI examination and subsequently to carotid US that has permitted the evaluation of IMT and IMTV parameters. From this data derives statistically significant correlation between IMTV value and WMH extension and the number of lesions preferentially located on the right hemisphere, while IMT did not show any significance or any predictive role for WMH’s evolution. These findings would support the hypothesis according to which IMTV could represent an early marker of endothelial dysfunction in the carotid artery and predictor factor of LA [40]. Another item to evaluate, according to increased cerebrovascular risks, is the structural composition of atheromasic plaque; in particular, fatty components is a critical factor for carotid artery vulnerability and increased risk of micro-embolization [41] associated with multiple infarct lacunas and WMH’s areas expansion as observed by Saba and colleagues. They planned a systematic evaluation of 50 patients, enrolled to Carotid-Endo-Arterectomy (CEA) who underwent firstly to CTA to well define stenosis degree, composition of carotid plaque and predict its behavior as stable or unstable on the basis of presence or not of fibro-calcific components; and then to brain MRI. The results showed a critical association between total carotid artery plaque volume and the onset of WMH areas mainly located in cerebral areas perfused by MCA in spite of Anterior (ACA) or Posterior Cerebral Artery (PCA) [42]. These behavioral differences relate to frequent variations and subsequently different compartmentalization of vascular blood-flow along Circle of Willis (COW) demonstrating lower risk of stroke and transient ischemic attacks among people who have effective collateral circles, while higher risk in those who have poor collateral circles [43].
On this basis, Saba et al., investigated about any existing relationships between COW variants and LA volume analyzing COW variations and WMH’s area by MRI on FLAIR and MRA on TOF sequences. The results confirmed not only a different location of LA distribution volume with predilection for MCA and PCA territories, but also a significant association between more COW existing variants and more WMH’s extension areas detected [44]. Bilateral occlusion of carotid artery, detected with CTA, would be determinant to emphasize a perpetual state of inflammation of blood brain barrier, through the activation in loco of MMPs that contribute to brain injury. In particular, the majority of the emboli, originated from principal atheroma, addressed through “perforating arteries” to most hypoxic sensitive territory such as hippocampus, white matter and some areas of cortex with a subsequent ischemia and genesis of WMH’s areas [45].
Further, Saba et al., analyzed, in a multicentric study, 98 patients who underwent CTA examination of brain and supra aortic vessels in parallel both brain white matter damage either grade of stenosis and nature of plaque. This study confirmed that CTA scan detect the major risk patient’s cohort developing a future LA and lead to consider white matter change as intermediate of stroke’s brain damage evolution in elderly vasculopathic patients [46,47]. All of these studies consider the presence or not of carotid artery stiffness [48], carotid artery wall thickness, composition and behavior of carotid plaque detected by echo-color Doppler, as independent risk factors for WMH genesis and distribution alongside the brain. In particular, patients with an unstable carotid plaque developed an extended distribution of WMH’s burdens especially on periventricular area, with associated cognitive decline [49].
Thanks to these findings, we can assume a new interpretative role of combined imaging (ECD, CT and MRI) as predicting the outcome in patient’s cohort undergoing to Carotid Endarterectomy (CEA). The global vascular impairment observed leads to negative influence and also to general outcome and successful recanalization of carotid vessel after CEA and Carotid Stenting Procedure (CAS) [50]. The most reasonable hypothesis is that basal hypo-perfusion leads to a reduced wash out of emboli not only on the treated artery side but also on the contralateral, leading frequently to ischemic brain injuries [51]. This feature explains how much the extension of WM lesions, detected by MRI with DWI and FLAIR sequences, could influence a clinical outcome and predict a higher probability of a new ischemic event after CEA among patients with asymptomatic ischemic brain lesions [52]. In the last decade, many studies focused on clinical impact of WMH; in particular, the European Leukoaraiosis and Disability (LADIS) study contributed to this porpoise substantially. This multicenter study provided harmonization between different diagnostic instruments (clinical, neuropsychological and radiological) to predict progression on global disability in subjects with a WMH load using Fazekas Scale and Scheltens Visual Rating Scale for global and temporal atrophy. After three years follow-up they found a significant correlation between moderate-severe WMH load and progression of clinical disability, such as speed and motor control, with annual transition of 29,5% [38]. They also suggested strong relationship between WMH load and cognitive decline as well as attention, visual-constructural and naming praxis depending from number and distribution of lacunar infarcts and depression [38]. Since depression can be described as a result of altered patterns of regulation between ventral structures (affective sphere) and dorsal structures (cognitive sphere), especially involving the prefrontal cortex, any vascular insults of this neural pathway can cause a disbalance and lack of modulation of limbic structures activity. Based on this hypothesis Aizenstein et al., conducted a functional imaging study using f-MRI among 33 elderly depressed patients and 27 nondepressed comparison subjects which demonstrated, among cohort of patients with middle and late-life onset of depression, a strong relationship between global cerebral WMH burden and higher degree of BOLD response of ventral and anterior structures (amygdala, rostral cingulate, prefrontal cortex and anterior insula) using faces and shapes affective reactivity task [39]. In particular, they observed in late-life onset of depression rostral cingulated hyperactivation associated with a greater WMH load as the strongest predictors of treatment despite middle-life onset of depression [39].
Corpus Callosum is another emerging important topic of study which can relate with cognitive and motor disabilities and many studies suggest that its involvement may be a stronger predictor of worst evolution on memory performance, executive dysfunction and speed [53]. As described above, WMH represents a very useful radiological “primer” to identify many pathological processes. Correct imaging assessment and scare present a crucial pathway to improve clinical approach predicting outcome and staging risk factors among their nature. Further perspectives on this field need many longitudinal studies are needed to evaluated effective long-term risk factors and predictors of clinical outcome.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
- Hachinski VC, Potter P, Merskey H (1987) Leuko-araiosis. Arch Neurol 44: 21-23.
- Sarbu N, Shih RY, Jones RV, Horkayne-Szakaly I, Oleaga L, et al. (2016) White Matter Diseases with Radiologic-Pathologic Correlation. RadioGraphics 36.
- Wardlaw JM, Valdés Hernández MC, Muñoz-Maniega S (2015) What are white matter hyperintensities made of? Relevance to vascular cognitive impairment. J Am Heart Assoc 4: 001140.
- Giwa MO, Williams J, Elderfield K, Jiwa NS, Bridges LS, et al. (2012) Neuropathologic evidence of endothelial changes in cerebral small vessel disease. Neurology 78: 167-174.
- Farrall AJ, Wardlaw JM (2009) Blood-brain barrier: Ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging 30: 337-352.
- Black S, Gao F, Bilbao J (2009) Understanding white matter disease: Imaging-pathological correlations in vascular cognitive impairment. Stroke 40: 48-52.
- Wardlaw JM, Smith EE, Biessels GJ, Cordonnier C, Fazekas F, et al. (2013) Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol 12: 822-838.
- Furuta A, Ishii N, Nishihara Y, Horie A (1991) Medullary arteries in aging and dementia. Stroke 22: 442-446.
- Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, et al. (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: A comparative histological study. Ann Neurol 30: 637-649.
- Erro ME, Gállego J, Herrera M, Bermejo B (2005) Isolated pontine infarcts: Etiopathogenic mechanisms. Eur J Neurol 12: 984-988.
- Duering M, Csanadi E, Gesierich B, Jouvent E, Hervé D, et al. (2013) Incident lacunes preferentially localize to the edge of white matter hyperintensities: Insights into the pathophysiology of cerebral small vessel disease. Brain 136: 2717-2726.
- Chabriat H, Vahedi K, Iba-Zizen MT, Joutel A, Nibbio A, et al. (1995) Clinical spectrum of CADASIL: A study of 7 families. Cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Lancet 346: 934-939.
- Birnbaum J, Hellmann DB (2009) Primary Angiitis of the Central Nervous System. Arch Neurol 66: 704-709.
- Abdel Razek AA, Alvarez H, Bagg S, Refaat S, Castillo M (2014) Imaging spectrum of CNS vasculitis. Radiographics 34: 873-894.
- Singh TD, Fugate JE, Rabinstein AA (2014) Central pontine and extrapontine myelinolysis: A systematic review. Eur J Neurol 21: 1443-1450.
- Pröbstel AK, Schaller A, Lieb J, Hench J, Frank S, et al. (2016) Mitochondrial cytopathy with common MELAS mutation presenting as multiple system atrophy mimic. Neurol Genet 2: 121.
- White WB, Marfatia R, Schmidt J, Wakefield DB, Kaplan RF, et al. (2013) INtensive versus standard ambulatory blood pressure lowering to prevent functional DeclINe in the ElderlY (INFINITY). Am Heart J 165: 258-265.
- Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, et al. (2003) From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: Part I. Circulation 108: 1664-1672.
- Wahlund LO, Barkhof F, Fazekas F, Bronge L, Augustin M, et al. (2001) A new rating scale for age-related white matter changes applicable to MRI and CT. Stroke 32: 1318-1322.
- Le Bihan D, Mangin JF, Poupon C, Clark CA, Pappata S, et al. (2001) Diffusion tensor imaging: Concepts and applications. J Magn Reson Imaging 13: 534-546.
- Etherton MR, Wu O, Rost NS (2016) Recent Advances in Leukoaraiosis: White Matter Structural Integrity and Functional Outcomes after Acute Ischemic Stroke. Curr Cardiol Rep 18: 123.
- Wintermark M, Sesay M, Barbier E, Borbély K, Dillon WP, et al. (2005) Comparative overview of brain perfusion imaging techniques. Stroke 36: 83-99.
- Brooks WM, Wesley MH, Kodituwakku PW, Garry PJ, Rosenberg GA (1997) 1H-MRS differentiates white matter hyperintensities in subcortical arteriosclerotic encephalopathy from those in normal elderly. Stroke 28: 1940-1943.
- Taheri S, Gasparovic C, Shah NJ, Rosenberg GA (2011) Quantitative measurement of blood-brain barrier permeability in human using dynamic contrast-enhanced MRI with fast T1 mapping. Magn Reson Med 65: 1036-1042.
- De Cocker LJ, Lindenholz A, Zwanenburg JJ, van der Kolk AG, Zwartbol M, et al. (2018) Clinical vascular imaging in the brain at 7T. Neuroimage 168: 452-458.
- Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA (1987) MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. AJR Am J Roentgenol 149: 351-356.
- Wahlund LO, Agartz I, Almqvist O, Basun H, Forssell L, et al. (1990) The brain in healthy aged individuals: MR imaging. Radiology 174: 675-679.
- Longstreth WT Jr, Manolio TA, Arnold A, Burke GL, Bryan N, et al. (1996) Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people. The Cardiovascular Health Study. Stroke 27: 1274-1282.
- Prins ND, van Straaten EC, van Dijk EJ, Simoni M, van Schijndel RA, et al. (2004) Measuring progression of cerebral white matter lesions on MRI: Visual rating and volumetrics. Neurology 62: 1533-1539.
- Caprio FZ, Maas MB, Rosenberg NF, Kosteva AR, Bernstein RA, et al. (2013) Leukoaraiosis on magnetic resonance imaging correlates with worse outcomes after spontaneous intracerebral hemorrhage. Stroke 44: 642-646.
- Saba L, Raz E, Bassareo PP, di Martino M, de Cecco CN, et al. (2015) Is there an association between cerebral microbleeds and leukoaraiosis? J Stroke Cerebrovasc Dis 24: 284-289.
- Lin Q, Li Z, Wei R, Lei Q, Liu Y, et al. (2016) Increased Risk of Post-Thrombolysis Intracranial Hemorrhage in Acute Ischemic Stroke Patients with Leukoaraiosis: A Meta-Analysis. PLoS One 11: 0153486.
- Bahrani AA, Powell DK, Yu G, Johnson ES, Jicha GA, et al. (2017) White Matter Hyperintensity Associations with Cerebral Blood Flow in Elderly Subjects Stratified by Cerebrovascular Risk. J Stroke Cerebrovasc Dis 26: 779-786.
- Lucatelli P, Montisci R, Sanfilippo R, Sacconi B, Suri JS, et al. (2016) Is there an association between leukoaraiosis volume and diabetes? J Neuroradiol 43: 273-279.
- Smith CD, Johnson ES, Van Eldik LJ, Jicha GA, Schmitt FA, et al. (2016) Peripheral (deep) but not periventricular MRI white matter hyperintensities are increased in clinical vascular dementia compared to Alzheimer's disease. Brain Behav 6: 00438.
- Grysiewicz R, Gorelick PB (2012) Key neuroanatomical structures for post-stroke cognitive impairment. Curr Neurol Neurosci Rep 12: 703-708.
- De Reuck J (1971) The human periventricular arterial blood supply and the anatomy of cerebral infarctions. Eur Neurol 5: 321-334.
- The LADIS Study Group, Poggesi A, Pantoni L, Inzitari D, Fazekas F, et al. (2011) 2001-2011: A Decade of the LADIS (Leukoaraiosis And DISability) Study: What Have We Learned about White Matter Changes and Small-Vessel Disease? Cerebrovasc Dis 32: 577-588.
- Aizenstein HJ, Andreescu C, Edelman KL, Cochran JL, Price J, et al. (2011) fMRI correlates of white matter hyperintensities in late-life depression. Am J Psychiatry 168: 1075-1082.
- Lucatelli P, Raz E, Saba L, Argiolas GM, Montisci R, et al. (2016) Relationship between leukoaraiosis, carotid intima-media thickness and intima-media thickness variability: Preliminary results. Eur Radiol 26: 4423-4431.
- Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, et al. (2003) From Vulnerable Plaque to Vulnerable Patient: A Call for New Definitions and Risk Assessment Strategies: Part II. Circulation 108: 1772-1778.
- Saba L, Raz E, Grassi R, Di Paolo PL, Iacomino A, et al. (2013) Association between the volume of carotid artery plaque and its subcomponents and the volume of white matter lesions in patients selected for endarterectomy. AJR Am J Roentgenol 201: 747-752.
- van der Zwan A, Hillen B, Tulleken CA, Dujovny M, Dragovic L (1992) Variability of the territories of the major cerebral arteries. J Neurosurg 77: 927-940.
- Saba L, Raz E, Fatterpekar G, Montisci R, di Martino M, et al. (2015) Correlation between Leukoaraiosis Volume and Circle of Willis Variants. J Neuroimaging 25: 226-231.
- Rosenberg GA, Wallin A, Wardlaw JM, Markus HS, Montaner J, et al. (2016) Consensus statement for diagnosis of subcortical small vessel disease. J Cereb Blood Flow Metab 36: 6-25.
- Saba L, Pascalis L, Sanfilippo R, Anzidei M, Bura R, et al. (2011) Carotid artery wall thickness and leukoaraiosis: Preliminary results using multidetector row CT angiography. AJNR Am J Neuroradiol 32: 955-961.
- Saba L, Sanfilippo R, Pascalis L, Montisci R, Mallarini G (2009) Carotid artery abnormalities and leukoaraiosis in elderly patients: evaluation with MDCT. AJR Am J Roentgenol 192: 63-70.
- Kandiah N, Goh O, Mak E, Marmin M, Ng A (2014) Carotid stenosis: A risk factor for cerebral white-matter disease. J Stroke Cerebrovasc Dis 23: 136-139.
- Raman MR, Kantarci K, Murray ME, Jack CR Jr, Vemuri P (2016) Imaging markers of cerebrovascular pathologies: Pathophysiology, clinical presentation, and risk factors. Alzheimers Dement (Amst) 5: 5-14.
- Maggio P, Altamura C, Lupoi D, Paolucci M, Altavilla R, et al. (2017) The Role of White Matter Damage in the Risk of Periprocedural Diffusion-Weighted Lesions after Carotid Artery Stenting. Cerebrovasc Dis Extra 7: 1-8.
- Caplan LR Hennerici M (1998) Impaired clearance of emboli (washout) is an important link between hypoperfusion, embolism, and ischemic stroke. Arch Neurol 55: 1475-1482.
- Ederle J, Davagnanam I, van der Worp HB, Venables GS, Lyrer PA, et al. (2013) Effect of white-matter lesions on the risk of periprocedural stroke after carotid artery stenting versus endarterectomy in the International Carotid Stenting Study (ICSS): A prespecified analysis of data from a randomised trial. Lancet Neurol 12: 866-872.
- Schmidt R, Ropele S, Ferro J, Madureira S, Verdelho A, et al. (2010) Diffusion-weighted imaging and cognition in the leukoariosis and disability in the elderly study. Stroke 41: 402-408.
Citation: Garofalo P, Suri HS, Porcu M, Suri JS, Saba L (2020) White Matter Hyperintensity and Vascular Disease from Biological Basis to Clinical Significance. J Non Invasive Vasc Invest 5: 017.
Copyright: © 2020 Paolo Garofalo, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.