Introduction
In industrialized nations such as the United States, the United Kingdom, and Germany, approximately 20% of the population has been reported to experience Adverse Reactions to Food (ARF) with wheat, nuts, fruits, and milk among the most common triggers [1]. Most ARF are not mediated by the immune system, such as lactose intolerance which is the most common ARF worldwide [1]. ARF may result in both gastrointestinal symptoms and/or extra-intestinal symptoms. Gastrointestinal food allergies (an IgE immune response) do exist in both children and adults but the majority of symptoms from ARF are due to non-immunologic reactions to foods [1]. Thus, although immunoglobulin testing of antibodies to food proteins (IgE, IgG, IgA, IgM) is of clinical value in identifying an adverse reaction, exclusive use of such testing may be limiting.Historically, the primary Wheat-Related Disorders (WRDs) have been identified as celiac disease (affecting approximately 1 in 100 in the general population) and wheat allergy (affecting approximately 1 in 1000 in the general population) [2]. After celiac disease and wheat allergy have been (largely) excluded, numerous Non-Celiac Wheat Sensitivity (NCWS) disorders may be considered. Recent studies and experimental data strongly indicate that NCWS exists in a substantial proportion of the population, that it is an innate immune reaction to wheat and that patients often present with extra-intestinal symptoms, such as worsening of an underlying inflammatory disease in clear association with wheat consumption [3]. A category of sensitivities referred to as non-celiac wheat sensitivity may include:• Non-Celiac Gluten Sensitivity (NCGS), whose published prevalence varies widely (0.5-13%) in western populations [4,5]• Wheat germ agglutinin sensitivity (a carbohydrate-binding protein that also functions as a natural pesticide) which has been associated with inhibition of gut epithelial cell repair and stimulating synthesis of pro-inflammatory cytokines and may have an antigenic impact on the intestinal epithelium at nano-molar concentrations [6,7]• Wheat Amylase Trypsin Inhibitors (ATI) which stimulate an innate immune response via activation of TLR4 on monocytes, macrophages and dendritic cells, leading to release of proinflammatory cytokines within 2-12 h [8]. Wheat ATIs are a family of up to 17 similar proteins of molecular weights around 15 kD and represent 2-4% of the wheat protein. With oral ingestion, they co-stimulate antigen presenting cells and promote T cell activation in celiac disease, but also in other immune-mediated diseases within and outside the GI tract [3]• FODMAPs (Fermentable Oligo-, Di-, Mono-saccharides and Polyols) which may primarily cause gastrointestinal symptoms through gaseous production and osmotic diarrhea and are present in many food products, with fructans (arabinoxylans) commonly present in wheat [9-12]• Exorphin sensitivity (the opiate receptor agonists gluteomorphins and prodynorphins in wheat) [13]
If not properly harvested and stored, wheat may also be infested with fungi such as aspergillus and fusarium, leading to consumption of harmful mycotoxins [3]. Thus, in looking for evidence of an adverse reaction to wheat, if one is looking exclusively for celiac disease or an IgE allergy to wheat, it is possible for a clinician to arrive at a false conclusion of safety.
Gluten and Celiac Disease
Gluten is made up of the storage proteins gliadin and glutenin, linked together by starch. In celiac disease, the Triticeae tribe of gliadin and glutenin proteins, including wheat, barley, and rye, triggers an immune response activated by dendritic cells in the proximal part of the small intestine. This leads to an inflammatory spectrum recognized first as small intestinal inflammation which then progresses through a degenerative process (Marsh Classification system 0-IIIa, b, c) to eventual total villous atrophy. CD is characterized by villous atrophy and positive serology for autoantibodies to tissue Transglutaminase 2 (tTG) and/or endomysium. Celiac disease is an autoimmune condition. It is characterized by autoantibody production (transglutaminase and/or endomysium), autoimmune enteropathy, and autoimmune comorbidities up to 30 times more prevalent than in the general population [14,15].Tissue Transglutaminase (tTG) autoantibodies detect CD with 95% sensitivity and specificity [16]. Tissue transglutaminase is an ubiquitous protein involved in a wide variety of cell processes and therefore has a role in wound healing, inflammation, autoimmunity, and others [17]. tTG is central to the pathogenesis of celiac disease. It potentiates the immunogenicity of gluten peptides in the small bowel through deamidation. Tissue transglutaminase is the main target antigen for EMA antibodies and tTG is therefore recommended as a primary screening test for CD [18]. In celiac disease, antibodies are also produced against endomysium, a structure of the smooth muscle connective tissue. Anti-endomysial antibodies show a sensitivity and specificity > 95% for detecting total villous atrophy CD, however its sensitivity is as low as 27% - 31% with partial villous atrophy [2,19,20]. The classical presentation of CD is diarrhea, steatorrhea, and weight loss, mainly reflecting the damage to the intestines. However it is more recently recognized as a multi-system disorder, affecting the nervous system, musculoskeletal system, cardiovascular system, skin, and liver [14]. There are many heterogeneous, non-specific symptoms of CD, making it difficult to diagnose and earning its epithet as a “clinical chameleon.” CD may manifest with gastrointestinal, heart, brain, liver, reproductive, or kidney symptoms [21,22].Therefore, primarily suspecting CD based on gastrointestinal symptoms alone is in error. Clinicians should be aware that wheat-related disorders can present with dramatically different clinical presentations from one patient to another varying from gastrointestinal, metabolic, musculoskeletal, neuropsychiatric, reproductive and skin disorders (Table 1). For every celiac patient with gastrointestinal symptomatology, there are eight with extra-intestinal symptoms [23]. In patients with recognized autoimmunity, it may be prudent to have an increased index of suspicion for a wheat-related disorder. Delayed diagnosis of CD leads to cases with serious, long-term complications, such as osteoporosis, psychiatric disorders, infertility and reproductive disorders, ataxia, seizures, and cancer. Atypical and silent forms of CD further add to the difficulty of reaching an accurate diagnosis [14].
Non-Celiac Wheat Sensitivity and Non-Celiac Gluten Sensitivity
While the antigenic specificity and pathogenic relevance of immunologic reactivity to gluten in celiac disease has been extensively researched, the immune response to non-gluten proteins of wheat has not been characterized. The main immunoreactive non-gluten antibody target proteins have been identified as serpins, purinins, α-amylase/protease inhibitors, globulins and farinins. Assessment of reactivity toward purified recombinant proteins further confirmed the presence of antibody response to multiple specific antigens [24]. The conundrum of non-allergy, non-celiac response to the ingestion of wheat has been explored in the literature for over 40 years. Originally it was labeled as non-celiac gluten sensitivity. However, because non-gluten components of wheat, barley and rye may induce the disease, the more inclusive term, “non-celiac (non-allergy) wheat sensitivity”, has been suggested [3]. NCGS and NCWS generally refer to a collection of non-specific symptoms in response to ingestion of gluten-containing cereals that resolve upon removal from the diet, in individuals for whom coeliac disease and IgE-mediated wheat allergy have been ruled out. A WRD may be associated with GI symptoms as well as numerous extra-intestinal symptoms [25]. There is now undisputable and increasing evidence for NCGS [4]. Its existence is well established in multiple double blind studies [26]. In a group of 200 patients referred to gastroenterologists for evaluation of a possible gluten-related disorder, 7% had celiac disease while the remaining 93% were classified as having non-celiac gluten sensitivity [27].Studies evaluating NCGS have generally been performed in adults, although pediatric cases have been reported [28]. In a prospective multicenter study (38 Italian centers-27 adult gastroenterology, 5 internal medicine, 4 pediatrics, and 2 allergy-all recognized as referral centers and included in the register of the Italian Health Ministry for the diagnosis of gluten-related disorders), performed over 1 year, 391 new cases of NCGS to 340 new cases of celiac disease were identified, giving a ratio of 1.15:1 [26]. NCGS can present with variable gastrointestinal and extra-intestinal symptoms, as shown in table 1. The most frequent gastrointestinal symptoms reported in the multicenter study were bloating and abdominal pain, found in 87% of females and in 83% of males. More than 50% of patients reported diarrhea, with the number of evacuations per day ranging from 3 to 10, while 27% had alternating bowel habits and 24% had constipation. After bloating and abdominal pain, epigastric pain was the most frequent symptom, being found in 52% of patients, followed by nausea, aerophagia, gastroesophageal reflux disease and aphthous stomatitis in decreasing prevalence [26]. The most frequent extra-intestinal manifestations of NCGS were tiredness and lack of wellbeing, reported by 64% (female) and 68% (male) of the enrolled subjects. In addition, a high prevalence of neuropsychiatric symptoms including headache (54%), anxiety (39%), 'foggy mind' (38%) and arm/leg numbness (32%) were recorded. Other extra-intestinal manifestations emerging from the analysis of the survey responses were joint/muscle pain resembling fibromyalgia (31%), weight loss (25%), anemia (due to both iron deficiency and low folic acid; 22%), depression (18%), dermatitis (18%) and skin rash (29%). None of these patients were positive for celiac disease [26]. Non-celiac wheat-related disorders may have higher rates of autoimmune processes, much like that found in celiac disease. In the most recent study to date comparing celiac disease, Non-Celiac Wheat Sensitivity and IBS, the incidence of a patient having elevated antibodies to self (ANA) with NCWS was 46% vs. 24% in celiac disease and 2% in IBS [29]. Although the pathophysiology of NCGS is being actively explored, the complete picture is still unknown. Gluten and glutenin peptides, distinct from those that initiate celiac disease, appear to trigger the innate and adaptive immune response in intestinal epithelia, dendritic cells, macrophages, and monocytes in NCGS [30-34]. It can present with intestinal and extraintestinal symptoms, yet standard serological tests are often negative, and genetic predisposition (HLA DQ2/8) is variable [4].
Diagnostic Testing
Historically, celiac disease was defined as total villous atrophy and therefore small intestinal biopsy has been acknowledged as the “gold standard” for diagnosis [35]. However, celiac disease, as is true of all autoimmune diseases, develops on a spectrum [36]. Total villous atrophy can be viewed as the “end stage” of an extended, activated immune response in the small intestine. As one of the following case studies will demonstrate, earlier (subclinical) stages in the development of autoimmunity provide a window of opportunity to address the activated, adaptive immune response before organ damage has progressed to the frank pathology and the diagnostic state [36]. Advances in diagnostic testing have yielded specific and sensitive serum tests for the detection of total-villous-atrophy CD: IgA tissue transglutaminase, IgA antiendomysial antibody, and IgG anti-deamidated gliadin peptide antibodies [14,35,37]. IgA tTG antibody is the most sensitive and specific serological marker of total villous atrophy CD [35]. Recent guidelines released by the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition state that high anti-tTG antibody titers is sufficient for diagnosis [38]. In adults, an IgA tTG three to five-fold above normal is diagnostic of total villous atrophy and a biopsy is therefore not required [39]. North American 2005 diagnostic guidelines recommend testing children with symptoms of celiac disease, or an increased risk of celiac disease, using serum tTG. Those with high serum levels are referred to a pediatric gastroenterologist for an intestinal biopsy and if abnormal histology is observed, the patient should be treated with a strict gluten-free diet [40]. In 2012, the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition reported that in a child with suspected celiac disease, a compatible HLA status, positive EMA, and tTG elevated more than 10-fold, the obligatory duodenal biopsy could be bypassed [41]. Wheat allergy is mediated by an IgE antibody response to allergens in wheat flour (such as gliadin, a-amylase/trypsin inhibitor, omega gliadins or a-purothionin), which trigger the release of histamine from basophils and mast cells [14,42]. Wheat allergy should be detectable with IgE antibody testing, although non-IgE-mediated wheat allergy does exist and may be indistinguishable from NCGS [43]. To identify the specific offending allergen, skin prick testing or serum IgE antibodies are good screening tests, but neither has optimal reliability. The sensitivity was 44% for skin testing and 56% for radioallergosorbent test, and the specificity was 67% for both tests [44]. Thus, screening for an immune-mediated ARF with a single category of immunoglobulin may not be adequate.Ninety to ninety-five% of patients with CD are positive for HLA-DQ2 and 5-10% are positive for HLA-DQ8 genes, however newer evidence shows 7% of those with celiac disease will not carry either of these two genes [21]. Historically, the human leukocyte antigens (HLA-DQ2 and/or HLA-DQ8) have been considered diagnostic prerequisites for celiac disease. A variety of genetic markers may indicate a vulnerability to the celiac spectrum. To date, approximately 54% of the genetics of CD can be explained by HLA plus 57 non-HLA single nucleotide polymorphisms (Figure 1) [45]. Previously referred to exclusively as “the celiac genes”, HLA-DQ2 and DQ8 are also found in 53% of NCGS cases [27]. Until recently, there have been no reliable biomarkers of non-celiac gluten sensitivity that have been universally accepted. Historically, compelling evidence identifies a useful clinical role for immunoglobulin testing (IgG, IgA, IgM) to multiple peptides of gluten [46]. Recently, Choung et.al., Have demonstrated novel sets of epitopes derived from gliadin which have a high degree of accuracy (99% sensitivity and 100% specificity, p < 0.001) in differentiating CD from controls, compared with standard serologic tests. This method of ultra-high-density peptide microarray is proving broadly useful in identifying an immune reaction to wheat outside of the standard antigen screen of a wheat-related disorder. The relative noninvasiveness, broad availability, and versatility of the high-throughput peptide microarrays make this technology well suited for incorporation into routine health care and also provide a promising new tool for biomarker discovery [47]. • Exorphin sensitivity (the opiate receptor agonists gluteomorphins and prodynorphins in wheat) [13].
Clinical Case Analysis
Here we describe three previously published, pediatric cases of wheat-related disorders (with and without enteropathy characteristic of celiac disease) whose clinical manifestations were widely disparate: type 1 diabetes, pediatric sclerosing cholangitis (liver failure), and conjunctival tumor with an assumed diagnosis of Kaposi’s sarcoma. The cases were selected specifically for the varied and serious clinical pathologies observed in children and the lack of “hallmark” gastrointestinal symptoms that would have normally tipped off the practitioner to a wheat-related disorder. Two patients had CD and one had NCGS. Organ systems affected were the pancreas, liver, and eye and pathology resolved with a gluten free diet. This review analyzes the similarities and differences in these three cases and serves as a clinical lesson about the varied and unexpected aspects of wheat-related disorders, with or without celiac disease. Reference to the existing clinical literature and underlying mechanisms are discussed.
Pediatric Type 1 Diabetes Mellitus without CD
Presentation and lab findings
Sildorf and colleagues describe a 6-year-old boy who presented with Type 1 Diabetes Mellitus (T1DM) without evidence of celiac disease or gluten sensitivity [48]. He was admitted to the hospital with polydipsia, polyuria, blood glucose of 255.6 mg/dL (normal, 65-99)*, ketonuria, and hemoglobin A1C of 10.4% (normal, <5.7%; prediabetes, 5.7-6.4%; diabetes, > 6.5%). He did not have diabetic ketoacidosis. His glutamic acid decarboxylase antibodies were positive. He was negative for islet cell antibody and insulinoma associated antigen-2A. Gliadin antibodies, human transglutaminase and endomysium antibodies were negative so he was given the diagnosis of T1DM without celiac disease (or any other wheat-related disorder).
Treatment and follow-up
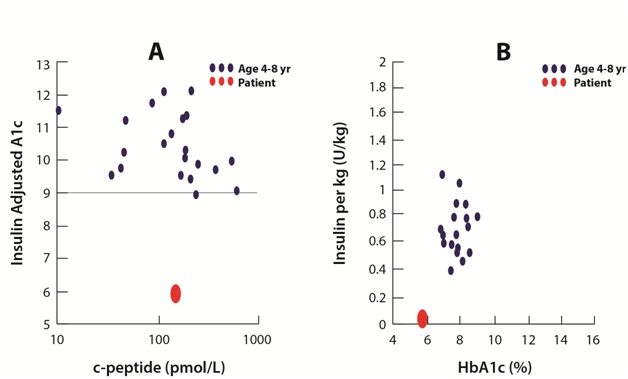
The patient was treated with insulin for five weeks (0.69 IE/kg/24h). Afterwards, blood glucose normalized so that the child was considered in the remission phase (72- 108 mg/dL)* and insulin was discontinued. HBA1c was 7.8%. Eight weeks post-diagnosis (three weeks after insulin treatment concluded), the family instituted a gluten-free diet as an adjunctive treatment with the hopes that it might preserve Beta cell activity. The clinic was preparing a study of a gluten-free diet intervention so it was offered to the child, even though he did not have celiac disease. The diet was also low glycemic, average calorie 7,085 KJ/day, and was split into six-seven small meals to be eaten throughout the day. Twelve weeks after diagnosis and four weeks after dietary changes, HbA1c was 6.7% (Figure 2). At eight months post-diagnosis, and after a challenge meal, C-peptide was 1.74 ng/mL* (normal 0.8-3.1) and proinsulin was 26 pmol/l (normal, 3-20 pmol/L). At 12 months post-diagnosis, C-peptide was 0.44 ng/mL* and proinsulin was 15 pmol/l. Unfortunately, stimulated c-peptide remained low and declined each month. However, insulin adjusted HbA1c is a surrogate marker for residual Beta cell function and remained low, suggesting the child was indeed in remission [48]. Remission was defined as either HbA1c < 7.5%, C-peptide above 0.91 ng/mL,* a daily insulin requirement < 0.5 units/kg/24 hr, or an insulin adjusted HbA1c < 9. GAD antibodies were not altered by the dietary changes. Sixteen months after diagnosis, serum fasting insulin was 0.66 uIU/mL* (normal, < 25mIU/L) and blood glucose was normal at 73.8 mg/dL*. Lipids were normal. Twenty months after diagnosis the patient still did not require insulin treatment. The authors stated in their conclusion, “At present time, the patient has been without daily insulin therapy for 20 months, a feature rarely seen in (diabetic) children of this age”.*Laboratory data has been converted to conventional units, including reference ranges, for a North American readership. See the original case report for data recorded in SI units [48].
Type 1 Diabetes Mellitus and Wheat-Related Disorders
Generally the diagnosis of T1DM precedes the onset of CD [49]. Ten percent of those diagnosed with T1DM will show evidence of celiac disease on duodenal biopsy [49]. In a large group of children with diabetes followed over six years, 60% were seropositive for celiac at the time T1DM was diagnosed and the remaining 40% were diagnosed with celiac four years later [50,51]. In adults with T1DM, 42% were found to have CD within 10 years of diagnosis and other studies show that serology is positive for CD 15 years after diagnosis [35]. While T1DM patients are twenty times more likely to have CD, the reverse is not true. Patients with CD are not more likely to have T1DM [49,51].The co-occurrence of celiac disease and T1DM are best explained by their shared genetic underpinnings, particularly the major histocompatibility complex class II antigen DQ2, encoded by DQA1*501 and DQB1*201 [52]. Other, non-HLA genetics have also been found in celiac disease and T1DM [35].The authors concluded that dietary changes, especially a gluten-free diet, led to a long, highly unusual, remission period of 20 months, without insulin therapy, in a child with T1DM. The child had a low C-peptide which suggested low levels of insulin production by the pancreas and higher risk of T1DM and elevated blood glucose. However, with dietary treatment, insulin sensitivity was enhanced and the C-peptide level and HbA1c levels remained stable. Blood glucose and insulin requirements normalized, placing the child in remission. To the author’s knowledge, this represents the first human trial of a gluten-free diet to effectively treat a patient with type 1 diabetes.There are mixed results as to the efficacy of a GFD in lowering hemoglobin A1c [35]. However, it poses no risks and may have helped to prolong remission and may have even preserved Beta cells in this case of T1DM [48]. The authors stated these learning points [48]:• Gluten-free diet prolongs remission• Gluten-free diet increases insulin sensitivity• Gluten-free diet is safe and acceptableUnlike the other case reports, the 6-year-old with type 1 diabetes was negative for human transglutaminase, endomysium, and gliadin antibodies and he was therefore negative for CD. Perhaps over 5-10 years, he would have eventually developed frank CD by standard diagnostic tests, as do other patients with T1DM. He fits the current criteria for non-celiac wheat sensitivity [43]. He never would have been trialed on a GFD except by happenstance. This raises the question: How many other children like him might derive benefit from a GFD, prior to developing serological indicators of a wheat-related disorder? Screening for a wheat-related disorder in patients with T1DM is advisable, though guidelines vary [49]. International guidelines call for screening of T1DM patients for CD at the time of diagnosis and then subsequently every five years, however the American College of Gastroenterology Guidelines and Canadian guidelines encourage CD screening only in the presence of suggestive symptoms [35]. However, symptoms are often absent and these criteria could therefore miss 40-60% of cases who have mild or absent gastrointestinal symptoms [35]. Classic celiac disease symptoms and histopathological findings are not sufficient to be considered exclusive indicators of celiac disease in children with T1DM and Down’s syndrome. Instead, authors recommend systematic serological screening because of the high prevalence of CD in these groups [50]. In type 1 diabetes, autoimmune disease destroys pancreatic Beta cells, causing permanent dependence on insulin and further negative complications affecting quality of life and life expectancy. If residual Beta cells can be preserved at the time of diagnosis, it can result in better short- and long-term outcomes [53]. When the onset of T1D is delayed, Beta cell function is better, there is a longer period of time preceding diagnosis, insulin autoantibodies are lower, patients are less frequently positive for HLA class II genes, metabolic function is preserved, and hA1C is lower at the time of diagnosis [54]. Early intervention with a GFD in those with wheat-related disorders could arrest or reverse disease processes and improve health throughout the lifespan. Pediatric Sclerosing Cholangitis (Liver Disease) with CD.
Pediatric Sclerosing Cholangitis (Liver Disease) with CD
Presentation and lab findings
An 11-year-old girl was referred to a Children’s Hospital for worsening liver failure [55]. She had suffered with fatigue, anorexia, and poor growth for three years. For one month prior she complained of jaundice and abdominal distention associated with mild itching. She had failure to thrive, being at the 3rd percentile for growth and height. She was pale, wasted, had clubbed fingers, and palmar erythema. She had splenomegaly and severe ascites. The liver was impalpable. She was negative for diarrhea, vomiting, fever, and had not taken herbal or medical drugs.Laboratory testing showed the following elevations: liver enzymes including ALT at 84 U/L, total bilirubin 98.7 umol/L (normal, 3-17), direct bilirubin 58.3 umol/L (0-3 umol/L), alanine aminotransferase was 200 (normal, 30-65 U/L). Gamma-glutamyltransferase was 111 U/L (normal, 7-32), clotting studies showed an international normalized ratio (INR) of 2.7, and albumin was 16 g/L (normal 35-50 gm/L). She was anemic with hemoglobin of 9.4 gm/L (normal, 11.1-15.9). Extensive laboratory testing for metabolic, endocrine diseases; viral hepatitis, Wilson disease, and alpha-1-antitrypsin deficiency were unremarkable. No other pertinent genetic tests were reported. Total immunoglobulin G was elevated at 23 gm/L (3.7-15). Celiac disease was suspected and tissue transglutaminase antibodies were measured at 385 units (normal, 0-20 units). Small intestinal biopsy showed total villous atrophy and crypt hyperplasia, characteristic of celiac disease. Ultrasound showed a coarse, shrunken, nodular liver with mild splenomegaly and gross ascites. There was beading and narrowing of the bile ducts consistent with sclerosing cholangitis. Liver biopsy revealed bridging fibrosis, portal tract expansion due to lymphocytes, plasma cells, and neutrophils. Bile duct proliferation and periductular fibrosis were also observed. She was diagnosed with Primary Sclerosing Cholangitis (PSC), a condition of swollen, inflamed bile ducts, leading to scar tissue and damage, inside and outside of the liver [56]. PSC is a chronic cholestatic liver disease of unknown cause, leading to inflammation and fibrosis of the biliary tree [57]. It results in end-stage liver disease, and is among the most common indications for liver transplantation in Europe and the United States, resulting in a shortened life span [58,59]. PSC significantly decreases survival. The median (50%) survival free of liver transplantation was 12.7 years [60].
Treatment and follow-up
The child was treated with ursodeoxycholic acid (20 mg/kg/day) and a gluten-free diet for life. She was prescribed prednisone (1 mg/kg/day) for three months which was then tapered down to 2.5 mg/day and discontinued at 6 months. After three months, liver enzymes normalized completely. The serum tissue transglutaminase antibodies were completely negative at 6 months. Four years later, liver function tests were normal and the tissue transglutaminase antibodies were still negative. However, abdominal ultrasound continued to show evidence of liver damage: a heterogeneous, coarse, shrunken and nodular liver. Serum bile acid and gamma-glutamyltransferase levels were normal. Liver autoantibodies were negative. Sclerosing cholangitis improved with a gluten-free diet, steroids (3 months) and ursodeoxycholic acid.
Liver Disease and Wheat-Related Disorders Commentary
Liver disease and histology in CD
Of the extraintestinal manifestations of CD, liver injury is very common [22], especially primary biliary cirrhosis and a mild, silent hepatitis often referred to as “celiac hepatitis” [61-63]. Hepatobiliary complications are seen in cases of gastrointestinal pathology, perhaps due to intestinal permeability brought on by the ingestion of gluten [61,64], contaminating portal blood flow [65], putting the liver in direct contact with nutrients, toxins, bacteria, lipopolysaccharides, inflammatory cytokines, and immunocytes from the gastrointestinal tract [64]. Liver abnormalities detected by biopsy in CD patients have been variable: no abnormality detected, non-specific hepatitis, or even severe changes such as fibrosis and cirrhosis [62]. The most common liver findings in celiac disease are isolated hypertransaminasemia with nonspecific histologic changes in a liver biopsy [63]. Hypertransaminasemia has been seen at the time of diagnosis in 40% of adults and 54% of children with classic CD [66-68]. However, only 9% of patients with CD have chronically elevated, unexplained, liver enzymes [68]. Hypertransaminasemia has been shown to completely resolve with a GFD in patients with CD [22,62]. Chronic, untreated CD may lead to severe liver damage such as cryptogenic chronic hepatitis or liver cirrhosis requiring liver transplantation [22]. GFD can help to avoid liver transplantation, resolve hepatitis, and even reverse liver damage [55,62]. A GFD leads to normalization of serum transaminases in 75% to 95% of patients with CD, usually within a year of good adherence to the diet (Table 2) [67,69,70]. All the patients with nonspecific changes in the liver histology and a follow-up liver biopsy normalized the histological changes after adherence to a GFD [66,70,71]. This is why reversible, wheat-related liver damage has been called celiac hepatitis [72].
Autoimmune liver disease
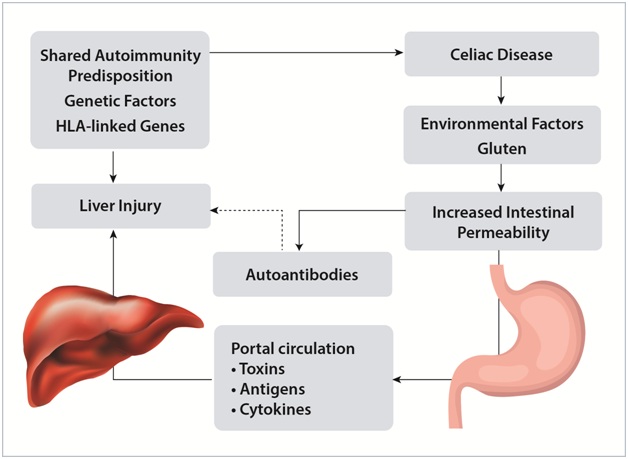
CD is associated with autoimmune liver diseases: primary biliary cirrhosis, autoimmune hepatitis, and primary sclerosing cholangitis. This association may be due to the HLA genes DQA1*0501 and DQB1*0201. Figure 3 shows one possible mechanism for the liver injury seen in celiac disease. Antinuclear antibodies and smooth muscle antibodies have been found in 20% to 60% of patients with autoimmune sclerosing cholangitis (as is the case in celiac disease), and could represent an immunopathogenic link between the two conditions [73].An association between CD and PSC was first described in 1988 [74]. A large, general population-based study from Sweden suggested that the prevalence of PSC increased 4- to 8-fold in patients with CD compared to those without CD [75]. Pediatric cases of CD with PSC have shown celiac disease to respond to a GFD, but not primary sclerosing cholangitis [74]. Given the severe liver damage seen in children with CD, researchers recommended that children with recent onset of celiac disease should be checked for liver function routinely and likewise, children with severe liver damage should be worked up for celiac disease [76]. According to Al-Hussaini and colleagues, only five other cases of pediatric liver failure due to celiac disease had been reported in the literature as of 2013 [55]. The etiology of sclerosing cholangitis is unknown. It is a chronic and progressive inflammatory disease [77]. Gradual autoimmune damage of the biliary tree leads to irreversible damage to bile ducts, persistent jaundice, ascites, varices, cholestasis, cirrhosis, liver failure, and death or liver transplantation within 10-20 years [56,73,77]. It is normally associated with inflammatory bowel disease and sometimes associated with celiac disease, chronic pancreatitis, diabetes mellitus, rheumatoid arthritis, or sarcoidosis [56,57]. Treatments are usually cholestyramine, ursodeoxycholic acid, fat-soluble vitamins, antibiotics for associated infection, and immune suppressant steroids. Surgical procedures are used to open up strictures of bile ducts in the liver and liver transplant is used for patients with end-stage liver disease [57]. There are no known treatments that bring about full remission from PSC [73,77]. Al-Hussaini and colleagues found that a gluten-free diet was the pivotal treatment for an 11-year old girl with liver disease and they concluded that it should be a part of routine screening in cases of unexplained liver disease. Because of the severity of the liver disease in this case, and unsure of the etiology, physicians treated this 11-year-old with prednisone and ursodeoxycholic acid, which are standard treatments for autoimmune sclerosing cholangitis and primary sclerosing cholangitis, respectively. However, pharmacotherapy did not prove to be necessary. Ursodeoxycholic acid and steroids do not lead to full resolution of disease or would have required continued treatment to produce long-term clinical relief [55]. In contrast, for this 11-year-old girl, prednisone could be fully discontinued six months later and all tests remained normal four years later, strongly suggesting that CD was the source of the liver disease. GFD did not reverse damage to the liver, as she “continued to show a heterogeneous, coarse, shrunken, and nodular liver” on follow-up. The authors suggested that “celiac-associated sclerosing cholangitis” might be an appropriate label for this case and that CD may be a treatable cause of liver failure [55].
Pediatric Conjunctival Tumor with CD
Presentation and lab findings
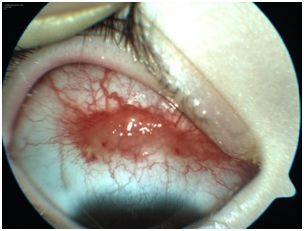
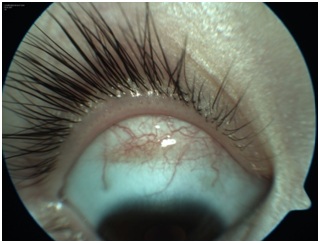
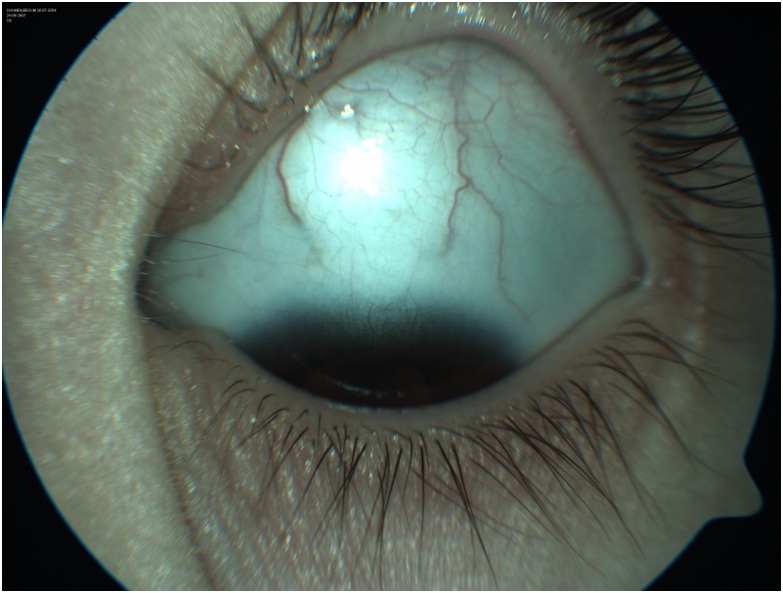
Tuncer et al., described a 3-year-old girl with a hemorrhagic conjunctival lesion on the right eye [78]. She had the reddish lesion for three months. It was not responsive to topical steroids and was referred for second opinion. Clinical history included early cessation of breastfeeding, intolerance to baby foods, abdominal distention for two months, and anorexia. She was below normal weight and height. She had recurrent serous otitis media (fluid in the middle ear) which had been treated with antibiotics, but the cause was unknown. She had undergone small intestinal biopsy due to suspicion of celiac disease at another clinic.The child had 20/20 vision in both eyes. But under examination by slit-lamp, there was a reddish, raised, “highly vascular spider-like lesion” on the superior bulbar conjunctiva of the right eye. It measured 12x4x2mm (Figure 4A). The presumed diagnosis was conjunctival Kaposi’s Sarcoma (KS). Kaposi’s sarcoma is a cancer that grows under the skin, and in the lining of the nose, mouth and throat. Abnormal patches of tissue may be red or pink and painful. Kaposi’s sarcoma is common in patients with immunosuppression, such as HIV and AIDS. However the child was negative for HIV. She showed elevated anti-gliadin and anti-endomysial antibodies (IgA). Intestinal biopsy showed partial villous atrophy, crypt hyperplasia, and intraepithelial lymphocytosis, diagnostic of celiac disease. The mother showed high levels of autoantibody titers for CD.
Treatment and follow-up
The patient initiated a gluten-free diet. No other protocols were initiated. After one week of GFD, the lesion gradually regressed. After two months on a GFD, the conjunctival lesion disappeared. At nine months follow-up the patient was completely asymptomatic and there was no recurrence of the ocular lesion.
The authors concluded: "We present a very unusual conjunctival tumor in a patient with CD that showed complete regression by a gluten-free diet. The precise pathological nature of this conjunctival lesion remains unknown due to the lack of histopathological confirmation. However, prompt regression of the conjunctival lesion during gluten-free diet suggests a possible relationship to CD and an autoimmune process".
Kaposi’s Sarcoma and Wheat-Related Disorders Commentary
A 3-year-old girl had a hemorrhagic conjunctival lesion diagnosed as Kaposi’s Sarcoma (KS), shortly before receiving the diagnosis of CD. Maintenance of a GFD led to spontaneous regression of assumed conjunctival Kaposi’s sarcoma [78]. CD was determined by intestinal biopsy. However, the diagnosis of KS was not confirmed by biopsy of the tumor because the parents did not want general anesthesia for their daughter [78]. The mechanism whereby CD led to Kaposi’s sarcoma in this 3-year-old is unknown. Authors concluded that CD let to an acquired autoimmune comorbid dysfunction, causing a lesion in the right eye with spider-like vascular extensions and subconjunctival hemorrhagic spots [78]. They postulated that anti-gliadin antibodies and/or circulating immune complexes deposited in the eye and affected the arrangement of capillary channels [78]. Neurological and psychiatric disorders have been noted in wheat-related disorders, suggesting that nerve tissue is damaged during the disease process [79,80]. In CD, gliadin antibodies may cross react with aquaporin, affecting the nervous system, the eye, and other tissues such as the gut [81]. Aquaporins are a class of cellular water channels found throughout the body [82]. Aquaporin channels may be thought of as a component of the “scaffolding architecture” inside the cell, especially in the nervous system. In the brain, they are responsible for water exchange, maintaining homeostasis, electrical activity, and neuronal signal transmission [83]. AQP4 is the most abundant water channel in the CNS, and is highly concentrated on the brainstem, spinal cord, diencephalon, hypothalamus, and optic nerves [82]. Molecular mimicry between gliadin peptides and aquaporin-4 may contribute to neuromyelitisoptica, an autoimmune inflammatory condition of the central nervous system, mainly affecting the spinal cord and optic nerves [79,84,85].
Discussion
These case studies show that a wheat-related disorder (with or without enteropathy) was related to the development of T1DM, liver disease, and a conjunctival tumor with a presumed diagnosis of Kaposi’s sarcoma, in three different children and a GFD arrested or reversed the disease processes. These cases demonstrate a glimpse of the varied clinical presentations, and varied pathogenesis, of wheat-related disorders in pediatric patients. Gastrointestinal symptoms, commonly believed to characterize celiac disease, were largely absent in all three patients.
Mechanisms of disease
Proteins in gluten can promote the initiation, development, and propagation of autoimmune disease, with or without celiac disease [14]. The classical paradigm of autoimmune pathogenesis involving a specific genetic makeup and exposure to environmental triggers has been challenged by the addition of a third element: the loss of intestinal barrier function [86]. This triad, or “perfect storm”, of genetic vulnerability, an environmental trigger (in the case of CD, gluten), and pathogenic intestinal permeability creates an opportunity for intervention before end-stage autoimmune disease develops [52]. It provides a leverage point where by re-establishing intestinal barrier function can arrest the autoimmune process and minimize the impact of genetic and environmental triggers [87]. The case of T1DM was likely due to an underlying autoimmune process triggered by wheat in take, even though gliadin, transglutaminase and endomysium antibodies were negative. This child qualified for the diagnosis of NCWS [3]. As gliadin sensitivity was negative, he may have been reacting to other peptides in gluten, not just gliadin. Poorly digested wheat may contain over 60 immunogenic peptides [88]. Non-gluten antigenic proteins found in wheat include serpins, purinins, α-amylase/protease inhibitors, globulins, and farinins [24]. The child was positive for GAD antibodies and remained positive even after a GFD [48]. Sclerosing cholangitis in the 11-year-old girl with CD may have also been an autoimmune process. Common, predisposing genetic factors (DQA1*501 and DQB1*201) may explain the increased prevalence of T1DM and liver disease in patients with CD and in the increased susceptibility of these children to these conditions. Gliadin initiates intestinal permeability in all individuals [65]. It is a proven cytotoxic protein, even for those without wheat-related disorders [4]. At the cellular level, it rearranges the cytoskeleton via zonulin, causes apoptosis, inhibits cell growth, changes the redox balance and enhances intestinal permeability by damaging tight junctions in the gastrointestinal lining [4]. Intestinal permeability may have therefore been a symptom in all three cases, although no testing was done to confirm this. The 3-year-old with a conjunctival tumor had a short duration of breastfeeding and multiple rounds of antibiotics for otitis media, suggesting microflora disturbance and vulnerability to pathogenic intestinal permeability. This could have contributed to the development of CD, accelerated vulnerability to, or exacerbated the condition. Antibiotics and reduced duration of breastfeeding have been shown to increase the risk of developing CD [89,90] and CD patients have abnormal microflora populations which continually stresses the epithelial lining in the GI tract [89,91]. Systemic damage due to CD may occur by a number of inflammatory pathways. In the intestinal lamina propria, gliadin triggers an increase in Intraepithelial Lymphocytes (IELs) which release inflammatory cytokines such as IFN-gamma, IL-6, and TNF-alpha. Additional macrophages and fibroblasts are recruited to the site leading to inflammation and destruction of the intestinal lining [35]. Higher levels of these cytokines and interleukins can be found in the systemic circulation of subjects with untreated CD. A GFD can lower the levels of these inflammatory markers and therefore the atherosclerotic and endothelial damage that they incur [35]. In CD, the number of gliadin-specific HLA-DQ2-restricted T-lymphocytes increases and antibodies against gliadin increase in circulation, potentially affecting the function of many organs [22]. Tissue transglutaminase-2 antibody affects endothelial cell adhesion and cell function and can cause vascular damage. tTG in liver diseases is associated with fibrogenesis, inflammation, and tissue repair. Patients with liver disease and celiac disease showed a higher expression of tTG in liver tissue [22].
Conclusion
These three pediatric cases of wheat-related disorders (with or without celiac disease) were chosen because they demonstrate widely disparate, serious clinical manifestations in children: liver failure, type 1 diabetes mellitus, and a conjunctival tumor with a presumed diagnosis as Kaposi’s sarcoma. The prognoses for these children, had they not been treated with a GFD, were poor. Autoimmune disease, complications, and even death could have followed. The child with type 1 diabetes would have had high HbA1cand high glucose (like his cohort). He would have likely developed full-blown type 1 diabetes and perhaps other autoimmune conditions over time. The child with primary sclerosing cholangitis would have progressed to liver failure and possibly death. It is reasonable to assume that the child with Kaposi’s sarcoma would have worsened and eventually developed symptoms of CD and/or other autoimmune comorbidities.Wheat-related disorders are systemic conditions. While there is increasing awareness that WRDs can manifest with extraintestinal symptoms, clinicians still rely too heavily on the presence of gastrointestinal symptoms to suspect a WRD. Patients with WRDs, who do not have gut symptoms, remain unidentified. The impact of a wheat-related disorder (with or without celiac disease) can manifest in any organ or system and has been shown to impact cardiovascular disease and malignant neoplasms, autoimmune diseases, connective tissue diseases, allergies, inflammatory bowel disease, nephritis, and even infections [22,35,92]. Previous antecedents (including stressors and exposures) and genetic susceptibilities can influence how gluten sensitivity manifests in the body. These cases showed wheat-related pathogenesis affecting the liver, pancreas, or eye, without notable gastrointestinal symptoms. These cases and the medical literature suggest that hypertransaminasemia, unexplained liver pathology, intestinal permeability, and autoimmune conditions, especially T1DM, warrant further investigation for an underlying wheat-related disorder.Classic symptoms of CD have been shown to poorly correspond with histopathological findings and are insufficient indicators of CD. Given that hundreds of protein interactions define a chronic disease, not a defect in any single pathway, a wheat-related disorder can present with a variety of symptoms due to its profound impact on the microbiome, and its systemic inflammatory and immunogenic impacts on the body [93]. These cases illustrate that “any symptom” in the body may be caused by a sensitivity to an environmental trigger (in this case gluten), initiating an inflammatory cascade. It is not restricted to one disease in one single organ system. For this reason, in patients with unexplained symptoms who do not improve with standard therapies, it may be prudent to screen for serological indicators of wheat-related disorders.
Acknowledgements
Special thanks to the authors of the case reports that made this analysis possible. Appreciation is owed to BMJ Case Reports, Hepatology, and the Indian Journal of Opthalmology for permission to reuse figures and tables.