Introduction
The global prevalence of infertility is approximately 15% with males accounting for 20-30% of case [1]. Factors that cause Male Factor Infertility (MFI) such as testicular injury, sexually transmitted disease and cancer can impair sperm function and integrity, affecting sperm motility, morphology and vitality [2,3]. MFI can also arise from congenital problems, including endocrinopathies such as androgen insensitivity syndrome [4] developmental disorders such as cryptorchidism [5], and chromosomal abnormalities such as Klinefelter syndrome or Y chromosome microdelections [6]. Sperm production and quality can also be influenced by aspects of lifestyle, such as diet and exercise, and environmental exposures to various compounds or xenobiotics [7]. As aberrant DNA methylation has been found to be more prevalent in infertile men, there is increasing interest in the epigenetic mechanisms behind these factors [8-12].
Epigenetics
Epigenetics refers to the heritable changes in gene function by mechanisms other than changes in the underlying DNA sequence. In contrast to traditional genetics in which mutation causes direct modification to the DNA sequence, epigenetics involves a complex set of regulatory machinery that function from the DNA to chromatin levels to influence gene expression, without affecting the fundamental nucleotide sequence itself [13]. Epigenetic control therefore, can modify a phenotypic outcome without affecting the genotype. This process is the fundamental driving force of variation in cell type and function in cells during differentiation from zygote to multicellular organism. Historically, this was illustrated in Conrad Waddington’s discovery of cell-fate determination by epigenetics [14], which revealed the importance of epigenetics in development. The molecular systems known to initiate and maintain the epigenome are DNA methylation and chromatin modifications including histone methylation and acetylation [13]. There is critical cross talk between this epigenetic machinery, which ensures correct gene expression and silencing in response to environmental cues [15,16].
DNA Methylation
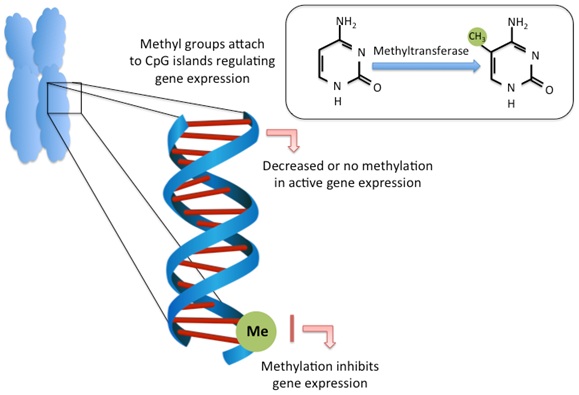
DNA methylation is the most well studied epigenetic mechanism in mammals [17]. It is a major regulator of gene expression and cell differentiation [18]. The capacity for DNA methylation marks to be stably maintained (i.e., somatically inherited) through cell division is the fundamental principle behind differentiation and the processes in which cells are able to interact with the environment. DNA methylation is the covalent attachment of methyl groups to DNA bases, occurring primarily on the 5’ position of cytosine bases in eukaryotic DNA. Cytosines targeted for methylation most often occur in CpG dinucleotides, where a cytosine precedes a guanine, although cytosine methylation can also occur at CpA, CpT and CpC sites [19]. CpG dinucleotides often cluster together as CpG Islands (CGIs) and are commonly found in the 5’ promoter region of genes where methylation suppresses and promotes gene expression, respectively [20-22]. DNA methylation is driven by the action of DNA Methyl Transferases (DNMTs), which convert cytosine to 5-methylcytosine (5mC) [23] (Figure 1).
The principle epigenetic modification of DNA in the mammalian genome is methylation of cytosine nucleotides. Methylation occurs mainly on the 5th carbon of the cytosine base, forming 5-methylcytosine (5-mC). DNA methylation is catalyzed by a family of enzymes called DNA methyltransferases and include DNMT1, DNMT3a and DNMT3b (box). Methylation mainly occurs around CpG clusters (CpG islands) at gene promoter regions is associated with gene inactivation.
CpG methylation at promoter or regulatory regions is typically associated with gene repression while lack of methylation promotes transcriptional activity [24,25]. Methylation causes transcriptional repression either through the direct blocking of transcription factor binding to DNA, or through methyl-CpG-binding protein recognition of methylated sites and their recruitment of co-repressors [26,27].
DNA methylation has important roles in embryonic development and differentiation [23], X-chromosome inactivation and imprinting [28], and suppression of transposable element activity [29]. However, these epigenetic processes are not just a developmental phenomenon; DNA methylation crucially continues to function throughout life responding to environmental cues to cause changes in somatic cell gene-regulatory programs. This epigenetic plasticity is influenced by numerous factors including age, lifestyle and environmental exposures that often lead to disease [30,31]. The gametes should be exempt from these types of epigenetic modifications but recent research suggests that some aberrant methylation marks may be maintained, which could have transgenerational implications for the health of future generations.
Epigenetic Reprogramming and Imprinted Genes
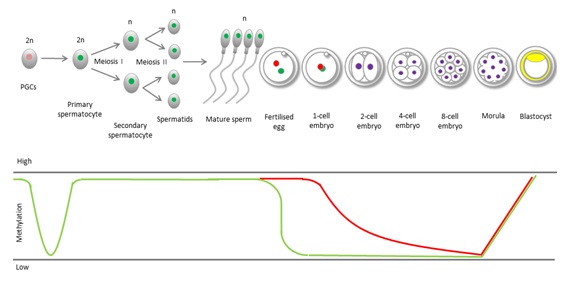
Epigenetic processes in gametes are different to those in somatic cells. During mammalian development, DNA undergoes two rounds of epigenetic reprogramming (Figure 2). The first is shortly after fertilization, where sperm DNA is stripped of the parental methylation signature. This global DNA demethylation occurs in sperm and oocyte DNA [32], and permits the totipotent zygote to initiate new gene transcription and de novo methylation in cells of the inner cell mass [32,33]. This allows expression and regulation of necessary genes important in the differentiation pathways leading to the specialization of cell types.
Primordial germ cells undergo rapid global demethylation after their migration into the gonadal ridge to remove parental imprints and allow establishment of gender-specific imprints. Another round of global demethylation occurs post-fertilization in the preimplantation embryo, where paternal DNA is immediately and actively demethylated (blue line) while maternal methylation persists until the 2-cell stage when it is passively removed (red line).
Despite this global demethylation of parental genomes, some genes escape this process and retain the methylation marks of the parental genome. These genes are known as imprinted genes, and are critical for embryonic development during the early rounds of transcription in the embryo. These genes show mono-allelic parent-of-origin expression and are expressed only from one parental allele [34]. The importance of this imprinting was illustrated in early pro-nuclei transfer experiments carried out by McGrath and Solter [35]. They found that when two haploid male pronuclei or two haploid female pronuclei were combined, although each genome was contributing half of the genetic information, there were sex-specific epigenetic marks corresponding to paternal or maternal DNA that were critical for embryonic development. Imprinting errors can therefore result in severe developmental disorders [36,37].
These imprints continue to persist in somatic cells after fertilization. However, developing gametes must undergo a second round of reprogramming to allow the establishment of new imprinting regions [28,36]. Primordial Germ Cells (PGC) have already acquired regional DNA methylation on their migration to the gonadal ridge (precursor to the gonads), similar to somatic cells [38], and demethylation therefore allows establishment of mature germ cell gender-specific genetic imprints [39].
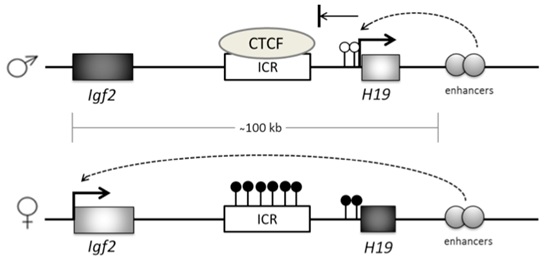
Imprinted genes are marked by Differentially Methylated Regions (DMRs) at CpG sites or CpG islands within or close to the gene itself [40]. Imprinted genes usually exist in clusters, and often the DNA methylation that controls the expression of these genes occurs at cis-regulatory sites, known as Imprinting Control Regions (ICR), whose methylation status can influence all genes in that cluster [41]. For example, a locus on chromosome 11 contains the IGF2 gene and H19 gene, which are expressed from the paternal and maternal DNA, respectively. Their expression is controlled by an enhancer downstream of H19 that is in turn, controlled by methylation at a DMR, which sits between the two genes [42] (Figure 3). When the DMR is not methylated, a zinc finger CCCTC-binding factor (CTCF) binds to the ICR and prevents enhancer-driven IGF2 activation, while allowing H19 activation. When the DMR is methylated, H19 transcription is repressed and the enhancer can interact with and activate IGF2.
Imprinted genes exhibit parent of origin gene expression. This is achieved through methylation-dependent gene silencing at Imprinting Control Regions (ICR). On the maternal allele at the IGF2-H19 locus the ICR is not methylated, allowing CTCF binding which silences IGF2 gene transcription through downstream enhancer activity. Maternally unmethylated H19 promoter (white lollipops) allows H19 transcription. On the paternal allele, CpG sites are methylated (black lollipops) on the ICR and in the H19 promoter region. This prevents CTCF binding, which allows enhancers to activate IGF2 while silencing H19.
An example of a serious congenital growth disorder that can arise from imprinting defects is in the IGF2-H19 locus of Beckwith-Wiedemann Syndrome sufferers. This condition occurs as a result of uniparental disomy, where both alleles are inherited from the father [43]. In this case the DMR is methylated on both alleles, leading to hyper activation of IGF2. As IGF2 is a growth promoter, the disease is characterised by serious fetal and neonatal overgrowth as well as a predisposition to tumour growth [44,45].
There are over 100 known imprinted genes in humans [46,47], most of which are involved in fetal, placental and brain development, post-natal growth, behaviour and metabolism [48]. This system ensures that a critical balance of gene dosage is maintained when parental chromosomes act together.
DNA Methylation in Sperm Function and Male Infertility
Once genomic imprints have been removed from the PGCs, spermatogenesis begins. A number of rounds of mitosis transform the PGCs into spermatogonia and primary spermatocytes, which then undergo two rounds of meiosis to become haploid round spermatids [49,50]. Finally, these spermatids differentiate into mature elongated sperm, which occurs simultaneously with remethylation of the sperm genome [51,52] (Figure 2). It is therefore probable that sperm abnormalities could be linked to aberrant DNA methylation during germ cell development, which is where much of the research in male infertility is focussed.
Most studies investigating the association between aberrant DNA methylation and infertility have focused on candidate imprinted genes [8-12]. As imprinted genes in germ cells have unique reprogramming events they are a central target for investigating aberrant methylation errors in sperm from infertile men. The importance of imprinted genes in post-zygotic gene expression also makes them a crucial objective, considering the implications of the transfer of incorrect imprints to the developing embryo [53] (Some imprinted genes associated with aberrant semen parameters are displayed in table 1).
Initial epigenetic studies comparing imprinted gene methylation in sperm DNA of oligozoospermic men (low sperm concentration), in comparison with normozoospermic men, (normal sperm parameters) found that the paternally imprinted H19 gene exhibited a loss of methylation and correspondingly the maternally imprinted Mesodern-Specific Transcript (MEST) gene had an increase in methylation [54]. The IGF2-H19 locus has been widely implicated in infertility and a number of DMRs exist in this locus. The DMR upstream of H19 contains a number of binding sites for the transcriptional repressor, CTCF, and is normally heavily methylated in sperm [55]. The 6th CTCF binding site has been identified as the most informative in infertile patients with severe sperm defects in morphology, motility and concentration, exhibiting hypomethylation compared with normal sperm [12,56].
Others have corroborated this methylation pattern seen in the H19 gene in further studies with oligozoospermic men [8,9,57]. Interestingly, Minor et al., found that sperm retrieved from the testes of men with obstructive azoospermia also demonstrated hypomethylation of H19, suggesting that aberrant methylation may be a product of the testicular environment and not singularly due to spermatogenesis failure [10].
Maternally imprinted genes have also exhibited increases in methylation, often corresponding to the decreases seen in paternally imprinted genes. MEST hypermethylation has repeatedly been linked to low sperm count, low progressive sperm motility and poor sperm morphology [8,61]. MEST is a candidate gene for the developmental disorder Silver-Russell Syndrome (SRS). Kagami et al., found that leukocyte DNA from a patient with SRS, was hypermethyled at the same CpG sites as in the paternal DNA in the MEST gene, suggesting that some imprinted genes may not follow the proper reprogramming stages during gametogenesis [55,62].
SNPRN is a maternally imprinted gene whose improper imprinting causes the developmental disorder Prader-Willi Syndrome (PWS) [63]. Loss of function of this gene on the paternal allele results in PWS. SNRPN was found to be hypermethylated in oligozoospermic patients as well as in patients with abnormal protamine ratios [58]. Other genes important for control of embryonic development and growth including PLAGL1, LIT1, MEST and PEG3 all showed hypermethylation in oligozoospermic patients in this study. Aberrant methylation of PLAGL1 has also been associated with poor semen parameters elsewhere [59], and proper methylation of this gene is critical for prenatal and post-natal growth in healthy infants [64].
Global DNA methylation does not appear to be a marker for infertility as many studies have found no difference between fertile and infertile males [9,11,12], indicating that gene-specific methylation errors are primarily susceptible.
Aberrant DNA methylation at imprinted genes has been widely associated with sperm abnormalities and infertility, as discussed. However, methylation at non-imprinted sites may also be important for expression of the paternal genome in the developing embryo. High throughput array technology has allowed the exploration of methylation across the whole genome, and has resulted in the finding that many non-imprinted genes are also linked to poor semen parameters [59,65].
Methylenetetrahydrofolate Reductase (MTHFR) is a major regulatory gene in folate metabolism. It is vital for providing sufficient methyl groups for DNA synthesis and methylation and is thought to have an important role in spermatogenesis due to its association with low sperm count and non-obstructive azoospermia [66,67]. MTHFR promoter hypermethylation has been strongly associated with idiopathic male infertility [62], and interestingly, this hypermethylation seems to occur alongside hypomethylation of H19 [69]. Given the role of MTHFR in normal methylation, this could have consequences for other genes. Not only has MTHFR hypermethylation been associated with male infertility, but also with spontaneous abortion rates [70], highlighting the importance of this gene for developmental processes.
Spermatogenesis genes are also susceptible to DNA modifications and can be directly linked to male infertility. Deleted in Azoospermia-like (DAZL) is a key spermatogenesis gene important for primordial germ cell formation, while it is also expressed in mature sperm [71,72]. Single Nucleotide Polymorphisms (SNPs) as well as aberrant methylation in the promoter of DAZL are associated with abnormal semen parameters [73,74], indicating that this is an important gene for male fertility. DAZL has been found to be abundantly expressed in the pre-implantation embryo from the 2-cell stage, however this is thought to come through maternal genome control, and so the contribution of paternal DAZL methylation is yet to be determined [74].
As DNA methylation is an important regulator of biological processes, including spermatogenesis, and given the occurrence of aberrant methylation in infertile men, it is possible that this epigenetic mechanism plays a role in male infertility.
Lifestyle and Environmental Effects on Fertility and DNA Methylation
DNA methylation in various tissues and cell types can be affected by factors such as age, stress, diet obesity, exercise and chemical exposures. Aberrant methylation in sperm DNA could be acquired at a number of important points during development, from as early as the reprogramming events in PGCs, and throughout the lifetime of the individual. Alternatively, some evidence suggests a possibility for epigenetic marks to be transferred through the paternal germline, where paternal sperm DNA methylation would bypass full reprogramming. The environmental and lifestyle factors contributing to sperm DNA methylation changes are discussed here, as well as the current knowledge on the inheritance of paternal methylation marks.
Age
The effects of male age on sperm quality and reproductive outcomes is an area of ongoing investigation. Significant reductions in semen parameters are seen with increasing age and paternal age is associated with reduced fertility, increased pregnancy complications and adverse childhood outcomes [75]. The mechanisms behind these issues have a genetic component, and may include factors that influence the epigenome.
DNA methylation changes with age, as illustrated by the divergence in genomic methylation between older monozygotic twins [76]. Global DNA methylation is generally found to decrease over time [77,78] while gene-specific methylation may be positively or negatively associated with age, and this occurs in a tissue-specific manner [79]. This is also the case for human sperm, as one study found both hypo and hypermethylation of a number of genes in sperm DNA of men analyzed 10 years apart [80]. It is possible that deregulated gene expression through changes in the epigenome could contribute to the age-related declines in fertility.
The role of paternal age in the development of offspring health is becoming an exciting area of research. Advanced paternal age can be a risk factor for some neurodevelopmental psychiatric disorders including schizophrenia and autism [82-84]. Changes in DNA methylation could be an underlying mechanism for some of these cases. For instance, Adkins et al., found differences in CpG methylation in newborns related to the age of the father, and some of these modified sites were found in genes that have been associated previously with schizophrenia and bipolar disorder [85]. Furthermore, Milekic et al., found that mice born to older males had DNA methylation abnormalities in the brain, which were similar to abnormalities seen in the paternal sperm of old mice but not in young mice. The off spring also exhibited behavioural differences that illustrate the effects of this aberrant methylation [86]. These studies indicate age-related DNA methylation changes in sperm may be retained in the embryo. However, to fully elucidate the mechanism behind the transgenerational effects of paternal age, more thorough longitudinal studies of offspring’s long-term health combined with DNA methylation analysis is required.
Obesity and nutrition
The global obesity epidemic is a result of a combination of genetic, epigenetic and environmental factors. It has arisen alongside an increase in the number of people seeking Assisted Reproductive Technology (ART) and amidst claims of decreasing male fertility [87-90]. Large cohort studies have attempted to identify whether obesity has a direct link to poor semen quality. Some have reported significant decline in semen parameters with increasing obesity, BMI and waist circumference [91-93], while others have found that obesity-associated markers are not good predictors of semen quality [93,94]. Mouse models have shown that induced paternal obesity can reduce fertilization capacity, as well as interrupt embryonic development. However, these may not truly represent the human scenario [96,97].
Obesity affects the methylation of genes in somatic cells regulating metabolism and satiety as well as cancer-associated genes [98-100]. Augmentation of DNA methylation and transmission of these marks to the next generation is important for understanding disease aetiology. Two studies have investigated DNA methylation in the offspring of obese fathers: the first found that the IGF2 DMR was hypomethylated in umbilical cord leukocyte DNA of the offspring from obese fathers [100]. Interestingly, this imprinted site is known to disrupt methylation in the blood DNA of obese adolescents [101]. The second study found that imprinted genes including MEST, PEG3 and NNAT, also demonstrated hypomethylation in offspring of obese fathers compared with non-obese fathers [102].
Mouse models have been an insightful platform for the study of the transgenerational epigenetic effects of nutrition on the development of health and disease in the offspring. Over feeding and starvation of male mice prior to mating can result in metabolic deregulation in their offspring [103,104]. Wei et al., demonstrated that induced paternal pre-diabetes led to altered methylation in the sperm DNA, as well as altered methylation and gene expression in glucose uptake and metabolism genes and insulin signalling genes in the offspring, leading to glucose intolerance and insulin resistance [105]. Furthermore, Fullston et al., fed male mice a high fat diet to induce obesity and found a reduction in sperm DNA methylation of 25%, and altered testis-specific gene transcription. The female mates and offspring were all fed a control diet, yet the F1 and F2 generations exhibited glucose intolerance and insulin resistance in males as well as obesity in females [106]. Further suggestion that this mechanism can occur through DNA methylation was demonstrated in the reduction in obesity levels in offspring whose obese pregnant mothers had a diet supplemented with methyl donors [107]. Although animal studies should be interpreted with caution, these examples show evidence for the non-genetic transmission of obesity and diabetes through the paternal germline, and as there is some evidence for transgenerational inheritance in humans, it is possible that DNA methylation is one of the fundamental mechanisms.
Smoking
Cigarette smoking is considered to be one of the major environmental modifiers of CpG methylation [108]. Smoking has been shown to reduce DNA methylation at multiple loci, and critically, these modifications can be partially reversed on smoking cessation [109]. However, smoking has not yet been linked to DNA methylation changes in sperm. It is well known that prenatal maternal smoking has serious implications for developmental gene expression, and consequently offspring health [110,111]. However, the role of paternal smoking in DNA methylation in offspring is less understood. Xu et al., found significant differences in gen-specific methylation in sperm DNA of mice exposed to cigarette smoke [112]. However, in humans paternal smoking was not found to be linked to altered H19 or IGF2 DMR methylation in umbilical cord DNA of newborns [113].
Some evidence suggests that paternal smoking does have implications for offspring health: reduced semen parameters have been detected in adolescents whose fathers were smokers, even though no difference was seen in the semen of the fathers [114]. In addition, the Avon Longitudinal Study of Parents and Children found that fathers who began smoking before the age of 11 years had sons with higher BMI compared to those who began smoking later in life [115]. This supports the thesis that male smoking can cause adverse health outcomes in future generations, through the male germline.
Environmental toxicants
The damaging health implications of environmental pollutants are well documented, and as air quality standards remain low in urban locations there is also concern for male reproductive health [116]. Correlative studies on occupational chemical exposures have been a source of information on the potential effects of chemicals on sperm. One study of men exposed to traffic pollutants through working at motorway tollgates found a decrease in total motility and forward progression in their sperm [117]. A recent report on pesticide exposure, measured directly through biomonitoring, showed a direct inverse correlation between consumption of pesticide residues on fruit and vegetables and sperm count, and the number of morphologically normal sperm [118].
These correlative analyses are supported in studies that measure physiological chemical concentrations in the semen of participants such as: urinary Bisphenol A (BPA) - a high volume chemical used in plastics manufacturing - was correlated with decreased semen quality and increased DNA damage [119]. Although endocrine disruption by BPA is likely to be one reason for perturbed sperm quality, one study in humans did find hypomethylation at the LINE-1 gene in men exposed to BPA compared with non-exposed men, which is indicative of a global reduction in DNA methylation [120]. Mouse studies, again, have shed light on the transmission of these epigenetic modifications through the male germline: IGF2 DMR methylation was perturbed in the F2 males of pregnant mice exposed to BPA resulting in metabolic dysfunction [121,122]. Animals exposed to vinclozolin and DDT have also demonstrated generational epigenomic modifications and associated diseases [123,124]. However, the extent of this epigenetic memory in humans in response to environmental chemicals is yet to be elucidated.
Imprinting Defects and Assisted Reproductive Technology
With 1 in 6 couples experiencing infertility at some stage in their lifetime, more people are seeking ART and the number of infants born through ART worldwide now stands at approximately 5 million [1]. This highlights the importance of ART as a standardised therapy for infertility. However, there has been an increase in the frequency of imprinting disorders, such as Beckwith-Wiedemann syndrome, in children born through ART [125,126].
ART involves exposing gametes and embryos to long periods of incubation in culture media or cryogenic storage. This artificial environment may result in modification to the gamete or embryo methylome. Ovarian stimulation has been linked to altered imprinted gene methylation in embryos [127,128]. Vitrified mouse oocytes have reduced methylation at imprinted genes in blastocysts, due to reduced DNA methyltransferase expression [129], and this was also found in a human oocyte cryopreservation study [130]. However, a study using human sperm cryopreservation noted no changes to methylation at imprinted genes, spermatogenesis-related genes or MTHFR in the sperm DNA [131]. Further investigations are required to fully confirm the effects of cryopreservation on the methylome.
Methylation marks from parental DNA could be passed down through the germline as IVF and ICSI bypass the normal selection processes, resulting in persistent parental epigenetic marks in the embryo. Kobayashi et al., found that imprinted gene methylation errors in concept uses conceived through ART could be directly associated with the paternal genome, but not in all cases [132]. In addition, although Lazaravicuite et al., found higher rates of imprinting disorders in ART compared with non-ART children; they could not identify any significant methylation differences at specific imprinted genes between these groups [125]. This lack of clear evidence for epigenetic variability could be due to the current methodology for determining CpG methylation, which is measured as the methylation in total DNA extracted from a purified sperm sample. However, a recent discovery of germline mosaicism in sperm indicates that current research could be overlooking intra-individual variability: single cell bisulfite sequencing showed that sperm of fertile men had homogenous methylation patterns at imprinted genes, while sperm of men with mild oligozoospermia had discrete groups of abnormally methylated cells [133]. This work demonstrates that individuals can have both correctly and aberrantly methylated sperm, which is important for ART procedures where sperm are selected, due to the potential transmission of these modifications through the male germline.
Conclusion
The role of DNA methylation in spermatogenesis, sperm function, infertility and transgenerational epigenetics is an exciting area of research.
The decline in male fertility over the past few decades has been the subject of intense investigation [89,134]. A high proportion of male infertility is of an unknown aetiology and it is likely that lifestyle and environmental factors play a significant role. Changes CpG DNA methylation levels can result from environmental and lifestyle exposures, which affect the regulation of gene expression altering sperm function and development of disease in the offspring [13,16]. The exciting question within this field of research resides over whether epigenetic modifications are transmitted and maintained through the germline, potentially harbouring damaging methylation marks for future generations. Moreover, as the use of ART increases, there is further potential for these modifications to be passed on to the developing embryo unchecked [132].