Introduction
Fanconi Anemia (FA) is a rare genetic disorder that is mainly inherited in an autosomal recessive pattern and is rarely X-linked. It was first described in 1927 by the Swiss pediatrician Guido Fanconi who described three affected siblings with congenital abnormalities and progressive marrow failure at the age of 4-5 years [1].
It is the most frequent inherited instability syndrome, characterized by bone marrow failure, hypersensitivity to cross-linking agents and high risk for Acute Myeloid Leukemia (AML). Ataxia Telangiectasia, Xeroderma pigmentosum and Bloom, Werner, Nijmegen, Li -Fraumeni and Seckel syndromes are also associated with inability to repair DNA damage [2,3].
Age of diagnosis ranges from birth to 49 years and the male-to-female ratio is 1.2:1, with a median age of 6,5 years in males and 8 years in females. The incidence rate is about 1 in 131,000 births in the USA; however the frequency is higher among specific populations due to founder effects [4].
FA patients exhibit hypersensitivity to agents that cause Interstrand DNA Cross-Links (ICLs), such as Mitomycin C (MMC), Diepoxybutane (DEB) and cisplatin, manifested as chromosomal fragility and radial formation that represent the biological hallmark of the disease. Chromosome instability is caused by defects in FA proteins which participate in several DNA repair pathways, including homologous recombination, DNA mismatch repair, nucleotide excision repair and translesion DNA synthesis [5,6].
Clinical Features of FA
Clinical features of FA patients include various congenital abnormalities, mainly skin and upper limbs disorders, (Table 1), progressive bone marrow failure and increased risk for squamous cell cancer [7-12]. However in 25-40% of patients the classical features may be very subtle or even absent [4,12,13].
Approximately 3/4 of FA patients develop marrow dysfunction, ranging from mild asymptomatic cytopenia to severe Aplastic Anemia (AA) within the first decade of life. Aplastic anemia usually occurs at a median age of 8-10 years. FA patients can develop neutropenia and thrombocytopenia as well as anemia, Myelodysplastic Syndrome (MDS) or AML [14,15]. The relative risk of AML as compared to the general population is much higher and the median age in reported cases is 13 years, with a range from birth to 50 years of age. There is a cumulative risk of more than 50% of marrow failure and 20% of AML by the age of 40. Absence of marrow dysfunction, however, does not rule out the diagnosis of FA [14-16].
Marrow cellularity is best evaluated by bone marrow biopsy and the bone marrow often appears dysplastic, but the relationship between clonal cytogenetic abnormalities and progression to leukemia is not always clear. The most common cytogenetic abnormalities observed in FA patients involve chromosomes 1,3,4 or 7, but there is a striking association between chromosome 3q26q29 amplifications (partial trisomies and tetrasomies) and rapid progression to MDS or AML [17].
Patients are also at high risk (30% by age 40) of developing specific solid tumors at an unusually young age, including head, neck, esophagus, liver tumors, gynecological squamous cell carcinomas of the lower genital tract and anal cancers [18-20]. Compared to the general population, the risk is approximately 50-fold higher for all solid tumors, but hundred- to thousand-fold higher for cancers of the head and neck, esophagus, liver, vulva, and cervix [18-21]. The mechanisms by which defects in the FA complex increase susceptibility to specific solid tumors are not completely elucidated. Genomic instability caused by subsequent somatic events occurring relatively early in the life of FA patients may explain the high rate of solid tumors in FA. It is also noteworthy that there are associations between oral androgens and liver tumors, as well as certain Human Papilloma Viruses (HPVs) and gynecologic, head and neck cancers [21-23]. The fact that approximately 25% of reported FA patients were diagnosed after developing cancer highlights the concern for early diagnosis of FA [18,22,23].
Genetics of FA
Sixteen genes associated with the FA phenotype have been identified since 1992. As table 2 shows molecular defects in the FANCA gene are the most frequent, followed by FANCC and FANCG. FANCB is a rare X-linked recessive gene, while all the others are autosomal recessive [24-29]. The highest frequency of molecular defects in the FANCA gene (80%) is found in Spanish Gypsies [30], while founder effects including mutations in FANCC exist in Ashkenazi Jews [31]. Alterations in FANCG gene are also present in high incidence in Portuguese-Brazilian, French Acadian, Korean/Japanese and South Africans [32].
Genotype/Phenotype correlation
FA cases caused by biallelic FANCD1/BRCA2 mutations are rare but are associated with an increased risk of medulloblastoma or Wilm’s tumor and other embryonal tumors that may precede development of aplastic anemia or an earlier onset of leukemia [33-38]. Children with biallelic PALB2/FANCN mutations tend to have more serious cancer-related phenotypes, with early-onset of Wilm’s tumor, AML and medulloblastomas [33-35,39,40]. Children with molecular defects in the FANCJ gene do not appear to have the severe phenotype associated with FANCD1/BRCA2 and FANCN/PALB2 [33-35,41].
Monoallelic mutations in FANCD1/BRCA2, FANCJ/BRIP1, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4 and FANCQ/XPF are related with ovarian and breast cancer, but molecular defects in these genes are also identified in the non-FA population [33,42-46].
FA Pathway
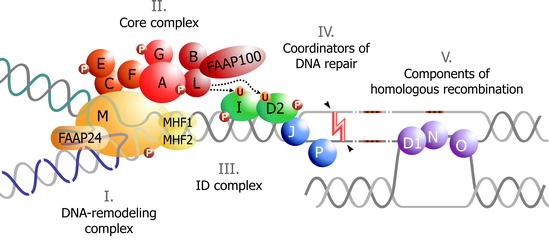
During the S phase of the cell cycle, DNA is duplicated at multiple replication forks. Interstrand Cross-Linking agents (ICL) prevent the separation of the two strands causing stalled replication forks and impairing DNA integrity [47]. The FA pathway responds to DNA damage, is involved in the process of DNA repair and the encoded proteins are grouped into 3 categories [48-51]:
- FA Core Complex (FA-CC) that includes eight FA-proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL and FANCM) and three FA associated proteins (FAAP20, FAAP24 and FAAP100).
- ID2 complex that includes the proteins FANCD2 and FANCI.
- Downstream proteins BRCA2/FANCD1, BACH1/FANCJ, PALB2/FANCN, RAD51C/FANCO, SLX4/FANCP, and XPF/FANCQ.
When responding to DNA damage FA-CC activates the monoubiquitination of FANCD2 and FANCI [51-53]. FAAP20 binds directly to FANCA and FANCG and participates in the regulation of the FA pathway [54]. FAAP24 interacts with FANCM and promotes the specific targeting of FANCM/FAAP24 to the binding sites of single-stranded DNA [55]. FAAP100 interacts with FANCB and FANCL to form a stable sub complex [56]. It is noteworthy that up to now, no mutation has been identified in FA patients in several genes encoding the FA pathway-related proteins, including FAAP20, FAAP24 and FAAP100 [54-56].
FANCL provides the E3 ubiquitin ligase activity of the FA-CC, which is necessary for the monoubiquitination of the ID complex [57]. Many of the FA-proteins are phosphorylated by the damage response kinases ATM and ATR. ATR is essential for the efficient monoubiquitination of FANCD2 and FANCI, implying an important role of ATR in the FA pathway activation [58,59].
The ubiquitinated ID complex is translocated to the chromatin at DNA damage foci at both sites and the surrounding areas [59]. The ubiquitinated ID-complex recruits endonucleases, mainly XPF/FANCQ-ERCC1, to ICLs [60]. The unhooking step of ICL repair is essential for resolving the ICL but creates a DNA Double Strand Break (DSB) that requires repair. CtBP Interacting Protein (CtIP) is shown to be a novel interacting partner of FANCD2 likely to promote DNA end resection and thus causing activation of HR or translesion synthesis, a repair mechanism that can bypass the ICL by creating a double stranded DNA molecule [61-63]. Downstream proteins have an important role in this repair mechanism, connecting the FA-pathway with DSB repair [48,49].
The remaining adducts are removed by Nucleotide Excision Repair (NER). The ERCC4 (FANCQ) gene encodes for a DNA repair endonuclease (XPF) that plays an essential role in NER and Interstrand Cross-Link Repair (ICLR) [60].
The FA-BRCA2 pathway is finally inactivated by the de-ubiquitination of the ID complex which is catalyzed by the USP1-UAF1 De-Ubiquitinase (DUB) (Figure 1) [64].
Diagnosis of FA
Early identification of FA allows differential diagnosis from other disorders, precludes inappropriate management of hematologic disease (such as AA, MDS, AML) and permits appropriate consideration of possible treatment or supportive care [4,7].
Cytogenetic analysis
The first diagnostic test used is the chromosomal breakage test. Two different sets of peripheral blood lymphocyte cultures - patient’s and control’s matched for age and sex- are set up and Mitomycin C (MMC) or Diepoxybutane (DEB) are added for clastogen-induced chromosome damage. As FA positive is considered the case in which the percentage of aberrations (breaks, gaps, rearrangements, radials, exchanges, and endoreduplications) in the patient’s lymphocytes is 7-10 times higher as compared to control (Figure 2) [5].
Molecular analysis
Following a positive chromosome breakage test molecular analysis is performed using either Multiplex Ligation-Dependent Probe Amplification (MLPA) [65], and/or sequencing, in order to identify deletions and/or point mutations in the 16 FANC genes [4,7].
If there is a founder effect in the population tested leading to a limited number of specific mutations there can be a targeted genetic diagnosis. These include Ashkenazi Jews (FANCC/FANCD1), non-Ashkenazi Jews Moroccan (FANCA), Tunisian (FANCA), Indian (FANCA), Israeli Arabs (FANCA/FANCG), Japanese (FANCC), Afrikaner (FANCA), Brazil (FANCA), Spanish Gypsy (FANCA) and Sub-Saharan African Black (FANCG) [4,30-33].
Flow cytometry
Flow cytometry examines cell cycle kinetics and can detect the proportion of cells that are arrested at G2/M after culturing in the presence of a clastogen, such as nitrogen mustard. With the exception of lymphocyte mosaicism, the cell cycle test reliably differentiates between FA and non-FA cases, including non-FA patients with aplastic anemia, Nijmegen breakage syndrome, Roberts, Baller-Gerold, VACTERL (Vertebral anomalies, Anal atresia, Cardiac defects, Tracheoesophageal fistula and/or Esophageal atresia, Renal & Radial anomalies and Limb defects), and other thrombo and erythropenia syndromes, as these conditions lack elevated G2-phase cell fractions. Flow cytometry can also be utilized to identify the FA complementation group using the appropriate antibodies for each subtype, or analyzing the correction of the characteristic FA-associated G2/M arrest after treatment of cells with DNA-damaging agents in combination with the complementation group analysis. This test is not used as widely as the chromosome breakage assay, however, and may give false negative results in FA patients with MDS or AML [66].
FANCD2 western blots
Following DNA damage, the complex of upstream FA gene products (FANCA, FANC B, FANCC, FANCE, FANCF, FANCG, FANCL, FANCM) leads to ubiquitination of the product of FANCD2, forming a longer protein (FANCD2-Ub), this can be distinguished from the shorter non-ubiquitinated one on a western blot using a FANCD2-specific antibody. FANCD2 protein monoubiquitination is essential for the functional integrity of the FA pathway as measured by resistance to MMC or DEB. This relatively inexpensive assay may be useful for the differential diagnosis of FA, but FA cases with significant lymphocyte mosaicism may be missed [52]. In addition the FA complementation group cannot be identified by this approach.
Complementation analysis
Patient’s lymphocytes or fibroblasts can be cultured with retroviruses which introduce known normal FANC genes in the patient’s cells, leading to the correction of the FA cellular phenotype (chromosome breaks or poor growth in the presence of a clastogen) depending on the location of the molecular defect on the FANC gene [67,68].
Next Generation Sequencing (NGS)
Since FA patients need special clinical management, diagnosis should be firmly established, to exclude conditions with overlapping phenotypes. NGS is performed by enrichment of the exons, flanking intronic and un-translated regions (5´ and 3´) of the specified genes using micro droplet PCR technology followed by sequencing with > 40 fold coverage at every target base. All pathogenic and novel variants, as well as variants of unknown significance as determined by bioinformatics, are confirmed by PCR and Sanger sequencing [69].
Multiplexed next-generation sequencing, based on massively parallel sequencing, is an effective molecular diagnostic approach for FA. The procedure, performed on genomic DNA, reduces the turnaround time, number of assays, and cost for a reliable detection of the disease-causing mutation. To increase the efficiency of the molecular diagnosis, genes involved in other bone marrow failure syndromes (e.g., Diamond-Blackfan anemia and Shwachman-Diamond syndrome) can be included, in order to diagnose these non-FA patients that are often referred for FA diagnosis [69].
Mosaicism in Fanconi Anemia
Approximately 25% of patients with FA show spontaneously occurring mosaicism with two subpopulations of lymphocytes, one of which is hypersensitive to cross-linking agents (MMC, DEB), while the other responds normally. Mosaicism, that may improve survival for the patients, can occur by mechanisms such as back mutation, intragenic crossover, gene conversion and compensating deletions/insertions. If lymphocyte analysis is inconclusive because of revertant mosaicism, fibroblasts are obtained since they do not show genetic reversion in vivo [70,71].
Prenatal Diagnosis and Genetic Counseling
Indications for prenatal testing include targeted prenatal testing when the molecular defect is known, ultrasonographic findings, or families with a prior affected child when no information is available on the molecular defects.
Chromosome breakage test
Prenatal testing is performed by cytogenetic analysis after the addition of DEB/MMC to CVS or amniotic fetal cells cultures in order to look for increased chromosomal breakage [72].
Molecular testing
The disease-causing mutations in families with an affected child must be identified before prenatal testing. Analysis of DNA extracted from fetal cells, obtained by Chorionic Villus Sampling (CVS) at 10 to 12 weeks of gestation or amniocentesis at 15-18 weeks of gestation, reveals the existence or not of the molecular defect. The techniques used are MLPA and Sequencing [73].
Fetal ultrasound evaluation
Ultrasound examination can be used to evaluate fetal anomalies consistent with FA. However, this is not a diagnostic test for FA and some characteristic congenital anomalies may not be detectable by ultrasound examination [74].
Preimplantation Genetic Diagnosis (PGD)
PGD may be performed in families where the paternal and maternal mutations have been identified and embryos selected do not have FA or are heterozygotes. In addition, single cell PCR allows combined PGD and HLA antigen testing [75,76]. PGD is an alternative prenatal diagnosis technique that offers the possibility not only to avoid the termination of a high risk pregnancy, but also to select embryos with particular genetic parameters that benefit an affected member of the family [77].
Genetic counseling: Genetic counseling of families with FA depends on the FA subtype and should include family and pregnancy histories, clarify the mode of inheritance and explain the genetic testing process [78]. It must provide information about current research opportunities, support groups and future reproductive options. Family members must also be informed about the possibility of developing ovarian and breast cancer if they are carriers of molecular defects that are located in the high risk FANC genes [79]. In case of novel molecular defects, further investigation is required in order to elucidate the consequences in DNA expression, since new approaches for treatment of FA patients may arise [78].
Treatment
There are short-term and long-term protocols for treatment of FA patients depending on the symptoms. Since the main disorder is the progressive dysfunction of bone marrow, treatment includes:
Androgens: Approximately half of FA patients respond well to androgens, which stimulate the production of red blood cells, platelets and sometimes white cell production. It is essential that use of androgens is considered in the context of an eventual bone marrow transplant, as their use may adversely affect the ultimate success of a transplant [80].
Growth factors: Growth factors (G-CSF) stimulate the production of white blood cells [80].
Bone marrow transplantation: This is the only long-term cure for the blood defects in FA and FA was the first disease for which cord blood transplantation was introduced [81].This treatment has many risks for FA patients because of their extreme sensitivity to radiation and chemotherapy [80,82].
New Insights
FA presents great heterogeneity including serious congenital and hematological abnormalities and researchers worldwide try to develop diagnostic tests, effective treatments and possibly a cure for this disease. New insights for diagnosis of FA are:
Fanconi anemia antibody project: The Fanconi Anemia Research Fund, through a partnership with Oregon Health & Science University, has established the Fanconi Anemia Antibody Project, which concerns the development of antisera against the FA complementation group proteins. The technique mainly used in this research protocol is western blot [83].
MicroRNAs: MicroRNAs (miRNAs) regulate gene expression post-transcriptionally, are involved in biological processes, such as cell proliferation, differentiation and apoptosis and are deregulated in cancer. The hypothesis that microRNAs regulate the FA-BRCA pathway and promote chemosensitivity and genomic instability in cancer cells has been proven; several microRNAs inhibit Ionizing Radiation (IR)-induced nuclear foci formation of FANCD2 and/or RAD51. Specific miRNAs are deregulated and play a role in the hematopoietic dysfunction of FA patients [84]. One of those, hsa-miR-181c, interacts with the 3 UTR of TNF, inhibiting its expression and toxic effects in hematopoietic progenitors from FA patients [85]. Studies of microRNAs may lead to the discovery of novel factors that are potential targets for chemo sensitization. These may be novel Fanconi anemia genes or breast/ovarian cancer susceptibility genes that are involved in the pathway and microRNA analysis may constitute a new diagnostic tool for Fanconi Anemia.
Aldehyde blocking agents: Endogenous aldehydes are an important source of genotoxicity in the human hematopoietic system, and the FA pathway counteracts them [86]. If the FA pathway is compromised, Hematopoietic Stem Cells (HSCs) accumulate aldehyde-induced DNA damage, resulting in BMF. Some modifier genes or environmental factors might affect levels of aldehydes or other genotoxic substances. This may be the reason why some ALDH2-proficient FA patients develop BMF early [86]. ALDH2 encodes an aldehyde dehydrogenase that participates in the oxidative pathway of alcohol metabolism. Administration of ALDH2 agonists, such as Alda-1 or a similar drug could be beneficial for ALDH2-proficient FA cases, stimulating the enzymatic activity of both the normal and variant ALDH2 [87].
Over the last two decades enormous efforts in the clinical and genetic field have changed the character of FA from a life-limiting disease to a chronic condition of highly variable severity. Methods of in vitro gene therapy might complement these approaches, mainly for FA patients with molecular defects in the commonly mutated FANC genes [88,89]. Gene therapy in this form will principally address hematological problems associated with FA [90]. In addition, patient and family support groups have an essential role in this context.