Introduction
Plasma citrate is maintained at a normal constant concentration in humans and animals. In humans the normal plasma citrate concentration is within a range of ~100-150µM. Exposure to conditions that tend to increase or decrease the plasma citrate beyond this normal range will trigger physiological, endocrinological, and metabolic responses that restore and maintain the normal plasma citrate concentration. If the plasma citrate concentration is sustained at a decreased or an increased concentration beyond the normal range, a hypocitricemia or hypercitricemia exists which is indicative of a pathophysiological status. Such conditions can impose clinical consequences (described below). The maintenance and regulation of plasma citrate and its physiological/pathophysiological/clinical implications remain largely unknown, speculative, and/or overlooked by most of the contemporary clinical and biomedical research community.
The intent of this review is to bring attention to the importance of normal citrate homeostasis. A background is presented of the existing information regarding the factors and mechanisms involved in the regulation and maintenance of citrate homeostasis. The pathophysiological and clinical consequences of dysregulated plasma citrate are described. As best that we can determine, this is the first review that describes and integrates these citrate relationships collectively in the context of the homeostasis of plasma citrate and its implications in medicine. Hopefully, this will stimulate interest and support for research, which are necessary to establish and understand the important role of citrate in the normal functional activities of the body; and the clinical consequences of disrupted citrate homeostasis.
The Normal Source and Removal of Plasma Citrate
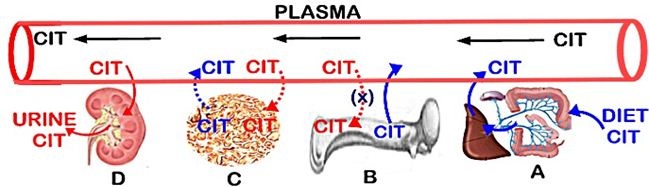
It is evident that there exists a balance between citrate that enters circulation and citrate that is removed from circulation, which results in the maintenance of the plasma citrate concentration. The prevailing views over time have included dietary citrate as the exogenous source, bone as the reservoir source of citrate, and tissue/cellular metabolism as an endogenous de novo source of plasma citrate. The removal of citrate from circulation has been considered to result from its urinary excretion, from its deposition into bone, and from its uptake and metabolism by various cells/tissues. However, much of the integrated role of these factors in the maintenance of plasma citrate has remained speculative and also inaccurate. The following provides a description of each of the factors as represented in figure 1, which leads to our current understanding of citrate homeostasis.
The sources of plasma citrate
Dietary citrate as an exogenous source of plasma citrate (Figure 1A): Depending on the amount ingested, citrate is absorbed from the digestive tract into hepatic portal circulation and ultimately into systemic circulation. The dietary citrate when available is an important source of plasma citrate. However, dietary citrate is not required for the maintenance of normal plasma citrate concentration. This is evident from the normocitricemia that exists despite the large variations in the citrate content of the diets of humans, which often contain minimal citrate. In animals with food supply containing minimal citrate, normocitricemia is well maintained. As such, other sources of citrate must exist, if or when the availability of dietary citrate is insufficient to maintain the normal plasma citrate.
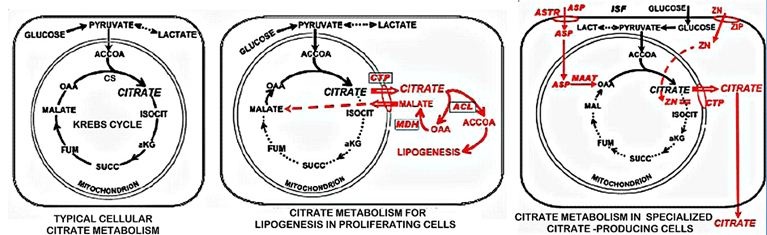
Soft tissue metabolic production of citrate as a source of plasma citrate (Figure 1C): All cells synthesize citrate as the initial metabolic step in the Krebs cycle (Figure 2). Typically in most cells, citrate is retained in the mitochondria for its entry into the Krebs cycle where it is utilized for energy (ATP) production and/or for the production of substrates for connecting metabolic pathways. Under some conditions, as in highly proliferating cells, the mitochondrial citrate is transported to the cytosol and utilized for lipid biosynthesis. In some specialized citrate-producing cells, the citrate is secreted from the cell into its extracellular compartment; such as osteoblast production and incorporation into bone. As such, soft tissue cellular metabolism does not provide a significant source of citrate into blood plasma.
Bone as a source of citrate (Figure 1B): Since first identified in 1941 [1], it has been established that bone in humans and all vertebrates contains extremely high levels of citrate [2]. About 90% of the total citrate in the body resides in bone; thereby constituting a major reservoir of citrate. Citrate is released from bone into plasma during bone resorption. This is the major source of citrate for maintaining the normal plasma concentration (described below).
The removal of citrate from blood plasma
Soft tissue uptake of citrate from plasma (Figure 1C): Since mammalian cells typically synthesize citrate as their metabolic source, they do not rely on the uptake of citrate from plasma. In addition, citrate in plasma exists as ~95% tricarboxylate---; ~4% dicarboxylate--; ~1% monocarboxylate-. As such, the plasma membrane of cells is highly impermeable to citrate, which prevents the cellular citrate uptake from extracellular fluid. Some cells, under special conditions will upregulate a plasma membrane citrate transporter (Slc13 family) for the uptake of plasma citrate. This is exemplified by kidney tubular cellular uptake of citrate from tubular fluid. Aside from such special conditions, the cellular uptake of citrate from plasma is not a significant factor in plasma citrate homeostasis.
Bone for removal of citrate from plasma (Figure 1B):
The prevailing view remains that citrate in plasma is “transported” with Calcium (CaCit) into bone (Figure 1B). However there has never been direct evidence of a process or mechanism for this purported CaCit “transport” from plasma to bone. Diffusion of CaCit from plasma to bone is highly unlikely since its concentration is much lower than in bone. Moreover, the osteoblast transport mechanism for calcium has not been identified; but likely occurs via a Ca++ channel, which would be independent of citrate transport. Also, the identification of osteoblast cell production of citrate in bone is independent of the transport of calcium; which, along with the absence of citrate transporter [3], make it highly unlikely that the removal of citrate from plasma and into bone is a relevant factor in plasma citrate homeostasis.
The renal clearance of citrate (Figure 1D): The kidneys remove citrate from plasma by the combination of urinary excretion of citrate and renal tubular cell uptake and metabolism of tubular fluid citrate; which together comprise the renal clearance of citrate. The renal clearance of citrate is the major factor for the removal of citrate from plasma and from the body.
The Mechanisms of the Role of Bone and Kidneys in the Maintenance of Plasma Citrate
Although the factors described above contribute to the concentration of citrate in plasma, the bone and the kidneys are the essential factors involved in the regulation and maintenance of plasma citrate homeostasis. The following describes the processes involved in their maintenance of the normal plasma citrate concentration.
Bone as the major source of citrate in plasma
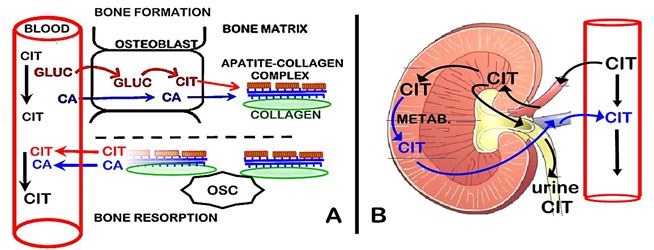
As shown in figure 3A, we recently identified that osteoblast metabolic production of citrate provides the source of citrate in bone [2-5]. During bone formation, the osteoblasts synthesize citrate, which is incorporated into the new bone. This occurs in concert with the osteoblast transport of calcium from plasma for mineralization in the new bone. These are separate but coordinated events that are required for the formation of the hydroxyapatite nanocrystal/collagen structure of normal bone.
During bone resorption, both citrate and calcium are released from bone into plasma. This provides a de novo source of citrate for the maintenance of the plasma concentration; whereas the calcium that enters the plasma is calcium that is originally derived from plasma (i.e., calcium turnover). Because bone turnover (bone formation↔bone resorption) occurs throughout life, it provides a continual source of new citrate as a major factor in plasma homeostasis. This is a new understanding of the important role of the osteoblasts and bone turnover in citrate homeostasis, which has major implications regarding the hormonal and other factors that regulate citrate homeostasis, and its clinical implications.
The renal clearance of citrate
The major removal of citrate from plasma is the renal clearance of citrate (Figure 3B). The glomerular filtrate derived from the renal artery initially contains the same concentration of citrate as exists in arterial plasma. When the glomerular filtrate enters the proximal nephron tubule some citrate is transported into the tubular cells. The citrate remaining in the tubular fluid is excreted as urinary citrate. In humans, the urinary excretion of citrate represents about 10-35% of the filtered citrate. Some of the citrate that is transported into the tubular cells is metabolized; and some citrate is reabsorbed back into the renal vein. Typically, the renal vein concentration of citrate is decreased by ~20-40% of the arterial concentration. Thus, the combination of the urinary excretion of citrate and the tubular cell uptake and metabolism of tubular fluid citrate constitute the renal clearance of plasma citrate. These relationships are described in the reviews [6-9].
Since normal renal function involves some level of obligatory citrate clearance (~500 mg/day), that loss of plasma citrate must be compensated by an equal addition of citrate into plasma. Dietary citrate, when available contributes to the replacement of citrate. However, bone resorption provides the citrate that is required to achieve the normal plasma concentration of citrate; and osteoblast de novo production of citrate refurbishes bone citrate during bone formation.
The Hormonal Regulation of Plasma Citrate Concentration
Exogenous and endogenous conditions and agents continually impose changes in the plasma citrate concentration. Such changes must be countered by adjustments in the citrate that enters the plasma and/or the citrate that is removed from the plasma to maintain the normal plasma citrate concentration. This requires endocrine monitoring of the plasma citrate concentration, and its hormonal response to maintain the normal plasma concentration; i.e., “citricemic” hormones. Unfortunately, aside from the early studies over the period of ~1950-1980, the focus on the physiological and endocrinological regulation of citrate homeostasis has been largely ignored. So, we must rely predominantly on the information provided in the early reports and integrated with the limited more recent reports as the basis for the relevant contemporary issues.
Parathyroid Hormone (PTH) and Calcitonin (CT); the major citricemic hormones
It has been well established that PTH and calcitonin CT are major respective hypercalcemic and hypocalcemic hormones. It is also well stablished that their calcemic actions are mediated mainly by their effects on bone and renal function. These relationships persist as the basis for contemporary clinical and biomedical research. Although the focus has been the calcemic role of PTH and CT, some early studies also identified PTH and CT as being respective hypercitricemic and hypocitricemic hormones. This relationship has been largely ignored, so that little progress has occurred in the identification of the mechanisms by which these hormones manifest their citricemic actions. As such, a prevailing view evolved and persists that the hormonal regulation of plasma citrate is coupled to the major calcemic effect as a “CaCit” effect. These issues are described below.
Parathyroid hormone:
Parathyroidectomy results in hypocalcemia and hypocitricemia; and PTH administration results in hypercalcemia and hypercitricemia. In addition, hypocalcemia and hypoctricemia result in parathyroid gland secretion of PTH to restore the normal concentrations of plasma calcium and citrate. PTH mediates its hyperciticemic action by its effects in bone and on renal function. PTH also mediates its hypercalcemic effect via its facilitation along with vitamin D of intestinal absorption and assimilation of dietary calcium. It is unlikely that citrate is coupled to that intestinal transport of calcium.
PTH effects on bone citrate:
The effect on bone results from PTH promotion of osteoclast bone resorption which releases citrate from bone to plasma (Figure 3A). In this regard, the citricemic and calcemic effects of PTH are “CaCit” coupled effects. It is also notable that early reports regarding bone metabolism effects of PTH consistently showed that PTH inhibits the oxidation of citrate, thereby increasing or conserving the citrate in bone [10-13]. However, the mechanism and implications of the PTH effect have never been established. Our recent studies with osteoblasts have demonstrated that zinc inhibition of citrate oxidation is essential for increased production of citrate (Figure 2) [3, 4], and this also applies to prostate epithelial cells [14]. The zinc/citrate bone relationship has important implications in bone disorders that exhibit decrease zinc and increased bone fractures (described below).
PTH effects on renal clearance of citrate: The effect on bone results from PTH promotion of osteoclast bone resorption which releases citrate from bone to plasma (Figure 3A). In this regard, the citricemic and calcemic effects of PTH are “CaCit” coupled effects. It is also notable that early reports regarding bone metabolism effects of PTH consistently showed that PTH inhibits the oxidation of citrate, thereby increasing or conserving the citrate in bone [10-13]. However, the mechanism and implications of the PTH effect have never been established. Our recent studies with osteoblasts have demonstrated that zinc inhibition of citrate oxidation is essential for increased production of citrate (Figure 2) [3, 4], and this also applies to prostate epithelial cells [14]. The zinc/citrate bone relationship has important implications in bone disorders that exhibit decrease zinc and increased bone fractures (described below).
Calcitonin: CT has been established and recognized as a hypocalcemic hormone (for reviews [21-25]). The release of CT from the thyroid gland into circulation results in hypocalcemia, as does the administration of CT. These effects are also manifested as a further decrease in plasma calcium in the presence of parathyroidectomy-induced hypocalcemia. Thus CT exhibits direct effects that are independent of the effects of PTH. In addition, hypercalcemia results in the thyroid secretion of CT for restoration of normocalcemia.
All of these hypocalcemia effects are accompanied by the corresponding CT hypocitricemia effects. Studies during 1970-1973 by Komarkova et al., [26] and Costello et al., [27,28] established the CT which induces hypocitricemia along with hypocalcemia. This was corroborated by Natelson et al in 1979 [29], who reported that administration of calcitonin in rabbits results in hypocitricemia. Since then, no other reports exist (as best we could determine) of the hypocitricemic effect of CT. Thus, a poor understanding of the mechanisms of CT regulation of plasma citrate and its pathophysiological implications currently exists. However, it is reasonably established that the citricemic effects are achieved by CT actions on bone and on renal citrate clearance, as are the calcemic effects.
CT effects on bone citrate: The persistent view has relied on the expectation that the major action of CT is its inhibition of osteoclast bone resorption, which decreases the release of CaCit into plasma and results in coupled hypocalcemia/hypocitricemia. This is supported by the effect of CT in increasing the release of citrate from bone into plasma [27]. However, the accompanying view that during bone formation, CT promotes CaCIT uptake from plasma which is incorporated into bone during bone formation. That is no longer tenable as an effect that manifests CT hypocalcemia and hypocitricemia.
The new understanding (Figure 3A) of osteoblast citrate production during bone formation and its independence of the calcium source, negates the CaCit coupling of the action of CT. Since CT exhibits direct effects on the osteoblasts during osteogenesis [30-34], it becomes extremely likely that CT promotes osteoblast production of citrate along with its mineralization during bone formation. Since no reported studies exist regarding CT effects on osteoblast citrate production, this is an essential issue that needs to be established.
CT effects on bone citrate: The persistent view has relied on the expectation that the major action of CT is its inhibition of osteoclast bone resorption, which decreases the release of CaCit into plasma and results in coupled hypocalcemia/hypocitricemia. This is supported by the effect of CT in increasing the release of citrate from bone into plasma [27]. However, the accompanying view that during bone formation, CT promotes CaCIT uptake from plasma which is incorporated into bone during bone formation. That is no longer tenable as an effect that manifests CT hypocalcemia and hypocitricemia.
The new understanding (Figure 3A) of osteoblast citrate production during bone formation and its independence of the calcium source, negates the CaCit coupling of the action of CT. Since CT exhibits direct effects on the osteoblasts during osteogenesis [30-34], it becomes extremely likely that CT promotes osteoblast production of citrate along with its mineralization during bone formation. Since no reported studies exist regarding CT effects on osteoblast citrate production, this is an essential issue that needs to be established.
CT effects on renal clearance of citrate: Our studies provide the only information of CT effects on the renal clearance of citrate [27]. Notably, CT treatment in rats results in ~500% increase in the urinary excretion of citrate. Since normal renal clearance of citrate includes ~10-35% urinary citrate, a 5-fold increase in urinary citrate would represent most or all of the renal clearance of citrate. This would imply that the tubular cell reabsorption of filtered citrate is limited by CT; which, if so, would suggest the absence of up regulated citrate transport. While this is speculative, the mechanism for this citraturic effect needs to be established. The citraturic effect is consistent with it being a major factor for the CT hypocitricemia in the hormonal regulation of plasma citrate.
It is notable that despite the focus on CT hypocalcemia, the effect of CT in the renal clearance of calcium remains conflicted. Some reports [35] conclude that CT results in increased renal clearance of calcium; other reports [36] conclude that CT results in decreased renal clearance of calcium. Since the latter is inconsistent with its being a factor in the hypocalcemic role of CT, it is presumed that the effect of CT on bone is the major action for CT hypocalcemia. Nevertheless, the renal handling of citrate is independent of calcium and a coupled clearance of calcium with citrate.
Do the citricemic hormones respond directly to the plasma citrate concentration? It is well established that PTH and CT are calcemic hormones that respond to changes in the plasma calcium concentration and promote the activities to restore the normal plasma calcium concentration. Citricemia consistently accompanies the PTH and CT calcemic response to the plasma calcium concentration; which has led to the view that the PTH and CT citricemias result from their regulation of CaCit. This is difficult to reconcile with the independent regulation of calcium and citrate during bone formation as represented in figure 3A. It is notable that Martin et al., [37] provided evidence that PTH regulation of citricemia was independent of its regulation of calcium. Nevertheless, the important issue of PTH and CT direct response to plasma citrate needs to be established.
There are conditions in which plasma citrate regulation exists independent of corresponding plasma calcium changes. For example, vitamin D promotes hypercitricemia, which involves bone and renal effects; whereas no corresponding effect on plasma calcium (and other parameters) occurs (described below). It is also notable that the administration of high levels of citrate results in a transient increase in plasma citrate and increased citraturia followed by a normocitricemia; without involvement of an accompanying calcemic response.
Surgical stress hypocitricemia: The most compelling evidence of an independent and direct citricemic effect in the absence of calcemic involvement is provided by the marked and consistent hypocitricemia associated with surgical stress in humans and animals [28,38,39]. By one day following surgical procedures, a marked hypocitricemia is established in patients and persists for several days; which occurs in the absence of corresponding hypocalcemia. In patients and in rats, the initiation of the hypocitricemia is not associated with an increased renal clearance of citrate. A marked hypocitricemic effect in the absence of a decrease in the plasma calcium was also exhibited following rectal surgery in a hyperparathyroid patient with hypercalcemia and hypercitricemia. The fact that the hypercalcemic and not the hypercitricemic effect of hyperparathyroidism is retained, provides evidence that PTH regulation of plasma citrate can be specific and independent of its calcemic effect. Thus the studies in humans and animals establish the existence of citricemic regulation that occurs independently of accompanying calcemic effects. The surgical hypocitricemia is not dependent on either PTH or CT involvement, or on adrenocorticoid. Therefore it results from a putative hypocitricemic hormone that has not yet been identified.
Hepatic Regulation of Plasma Citrate The prominent association of the liver with the circulatory system provides its potential impact on the plasma citrate concentration. We have noted in several reports the referral to an important normal role of liver clearance of citrate in the maintenance of normal plasma citrate concentration. However, the studies that established this relationship most often are not cited. Since the results of our studies described below are in conflict with that relationship, we conducted an extensive search for reported studies of liver clearance of plasma citrate. We found only three reports: one report using isolated liver perfusion studies that showed the clearance of citrate [40], another such study that showed the absence of liver clearance of citrate [41]; and another report that employed >100-fold higher than normal plasma citrate concentration, which purportedly showed liver clearance of citrate [42]. These reports, none of which employed physiological in situ liver studies, are insufficient to establish any functional role, or its absence, of the liver in the clearance of plasma citrate under normal conditions.
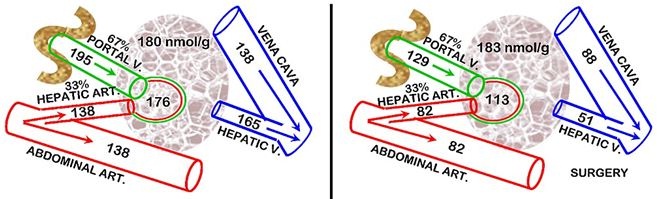
In our studies in humans and rats [38,39], we observed that the surgical stress hypocitricemic response did not involve any increase in the renal clearance of citrate; and it did not involve either PTH or CT. Consequently, we pursued the possibility of the liver clearance of plasma citrate being involved in the surgical stress hypocitricemia in rats. For this, we determined the hepatic Citrate Extraction Coefficient (CEC). The liver receives its blood supply from the Hepatic Artery (HA) and the Hepatic Portal Vein (HPV). In humans, HA provides ~25% and HPV~75% of the liver blood supply. In rats, the values are ~33% and 67%, respectively. Therefore the CEC equation for rats requires the calculation of the citrate “Arterial” concentration = (0.33HA) + (0.67HPV); then
CEC = (“Arterial” conc + Hepatic Vein conc) / “Arterial” conc”
These conditions (Figure 4) demonstrate that the liver citrate extraction coefficient is negligible in the normal animals; whereas surgical animals exhibit a CEC~0.55. Unlike the earlier reports, this study employs in situ liver citrate extraction from circulation which establishes the minimal, if any, normal hepatic citrate extraction. Thus, surgical stress induces a major hepatic citrate clearance of 55% of the citrate in the plasma that it receives. In the absence of increased renal clearance of citrate, the increased hepatic clearance of citrate is the major factor that results in surgical stress hypocitricemia. This becomes evident when comparing this hepatic citrate clearance to renal citrate clearance which is generally considered to be the major factor in the removal of citrate from circulation. Our rat studies showed that the normal renal citrate extraction is ~25% as compared to the liver citrate extraction of ~55% following surgery. The relative blood flow for the kidneys versus liver is ~1:1.4. Therefore liver citrate clearance following surgery is ~3-fold greater than normal renal citrate clearance. It also becomes evident that this high hepatic citrate clearance provides an explanation for surgical hypocitricemia over-riding the hypercitricemic effects of excessive parathyroid hormone and the hypocitricemic effects of calcitonin. This is an important liver relationship that had never been recognized.
These results also reveal that the hepatocytes in surgical stress animals must have an upregulated citrate transporter that facilitates the uptake of citrate from plasma; whereas under normal conditions the hepatocytes exhibit minimal citrate uptake from plasma. Also, the increased citrate extraction does not result in any increase in the liver tissue concentration of citrate; which demonstrates that the citrate is rapidly metabolized by the hepatocytes. These changes in hepatocyte metabolic activities must result from a surgical stress induced citricemic endocrine hormone response. Since PTH, CT, and corticosteroid are not involved, the putative hypocitricemic hormone remains unknown; and needs to be identified.
It is apparent from this study that the liver, under normal conditions is not significantly involved in the clearance of plasma citrate for the homeostatic maintenance of plasma citrate concentration. However, under specific conditions the hepatic clearance of citrate and the activities of the hepatic cells can be a major factor in maintaining the normal plasma citrate concentration.
The Pathophysiological and Clinical Consequences of Disrupted Plasma Citrate Homeostasis
The homeostasis of plasma citrate concentration must be essential for manifestation of important physiological relationships in the normal individual. Therefore, dysfunctional plasma citrate regulation should have pathophysiological and clinical consequences. However, the actual and potential implications of plasma citrate regulations are still not well established. Nevertheless, the following presents some important implications of dysfunctional regulation of plasma citrate concentration as represented by hypercitricemic and hypocitricemic conditions.
The plasma citrate-plasma calcium connection
Plasma calcium is associated with important physiological effects such as blood clotting, neuromuscular irritability, cardiac activity, and other effects. The effects are dependent on the total concentration of plasma calcium, on the molecular forms of calcium complexes, and on the concentration of Ca++. These have some dependency on the plasma citrate concentration. As such, changes in the plasma citrate concentration can impact the normal physiological effects of calcium; thereby resulting in pathophysiological and clinical consequences, including death.
An important example is the clinical consequences of the use of citrated blood for massive blood transfusion and blood exchange in adults and children. During the period of ~1945-1965, the introduction of massive blood transfusion as a medical procedure resulted in the identification of the clinical entity characterized as “citric acid (citrate) intoxication” [43-45]. The condition is a severe hypercitricemia (plasma citrate concentration is increased by ~10-fold or greater); which decreases the Ca++ by ~50% or more. The consequences included impaired blood coagulation; extended bleeding time, impaired cardiac function and EKG, hypotension; tetany; metabolic alkalosis, and in some cases, death. Since then, the issue of “citric acid intoxication”, as indicated by the sharp decline in published reports, had not received much attention. The recent increasing applications of massive blood transfusion for organ transplants, major surgical tissue resections, and for blood exchange has renewed the interest in the issue of citric acid intoxication, as is evident from recent reports such as [46-48]. The potential for citric acid intoxication is reflected in the conclusion [44] that “it is possible to predict with assurance that citric acid intoxication is likely to occur during multiple transfusions of citrated blood in patients during extremely rapid and prolonged infusions of citrated blood or plasma”. The implications of citrated blood for pediatric as well as adult conditions warrant its consideration and management.
The plasma citrate; renal clearance of citrate; calcium stone formation
During normal conditions, renal tubular fluid calcium is complexed with citrate as the soluble CaCit salt, which reduces the Ca++ concentration. This limits calcium super saturation, prevents nucleation of calcium oxalate and calcium phosphate; and the citrate also prevents crystal agglomeration and growth. Thus, hypocitraturia (generally less than ~320 mg citrate/day) is associated with 20-60% of cases of stone formation. This renal clearance of citrate relationships are described in the following reviews [6-9].
Conditions that decrease the concentration of citrate in the tubular fluid will lead to hypocitraturia and stone formation. This includes hypocitricemia in which the concentration of citrate entering the glomerular filtrate limits its availability in urine formation. Hypocitricemia due to insufficient dietary source of citrate can be such a factor.
However, hypocitraturia often occurs in the presence of normocitricemia and hypercitricemia. This is represented by acid–base status, which is the most important determinant of urinary citrate excretion. Metabolic and cellular acidosis is a major cause of hypocitraturia. Acidosis increases the transport of citrate from the tubular fluid into the tubular cells, and it increases the cellular metabolic utilization of the citrate. Consequently, urinary citrate is markedly decreased. Conditions that lead to metabolic acidosis or cellular acidosis (e.g., potassium deficiency, excess sodium, ketosis, exercise) will promote hypocitraturia.
A different effect is exemplified by hyperparathyroidism as a cause of hypercalcemia along with hypocitraturia and stone formation. In this case, the renal clearance of citrate is decreased; and the reabsorption of citrate from the tubular fluid back into circulation is increased. This differs from acidotic hypocitraturia, in which the reabsorption of citrate back into circulation is decreased. This difference implies that tubular cell uptake of citrate occurs in both conditions, but the cellular utilization of citrate is inhibited in response to parathyroid hormone (4.1.1b above).
The complexity of interacting factors in the renal clearance of citrate mandates the need for continued research into these issues.
Citrate dysregulation and bone disorders
The incorporation of citrate into the structure of bone is essential for optimal manifestation of the biomechanical properties of bone; e.g., stability, strength, and resistance to fracture [2,49,50]. Conditions that cause hypocitricemia can lead to the loss of bone citrate, and bone disorders that are characterized by loss of strength and increased bone fractures. This is well represented in vitamin D-deficient rickets, which is accompanied by hypocitricemia, and is treatable, by increased citrate availability (described below). Osteoporosis and other bone disorders that exhibit bone fragility and fractures are likely to have loss of bone citrate; especially if accompanied by deficient zinc, which is essential for osteoblast citrate production 9 (Figure 2 and Figure 3A). In an osteoporosis study, Cauderella et al., in 2003 [51] reported a stepwise logistic regression study “to find the biochemical parameter which related best with vertebral fractures: and citrate showed the highest statistical significance (p = 0,001)”. This would explain the success of the administration of zinc supplement, calcitonin, and vitamin D for restoring bone strength and decreasing bone fractures in osteoporosis. The contemporary clinical community must recognize the implications of citrate in these bone disorders.
Vitamin D and the Maintenance of Plasma Citrate Concentration
Although not a hormonal factor that directly responds to the plasma citrate concentration, there are numerous reports [51-56] that describe the role of vitamin D in the maintenance of the normal plasma concentration. Vitamin D deficiency results in hypocitricemia and administration of vitamin D increases the plasma citrate concentration. Along with the increase in plasma citrate, vitamin D also increases kidney tissue and bone citrate concentrations. The citrate effects of vitamin D are not dependent or associated with calcium or phosphate status, alkalosis, or other such conditions; thereby demonstrating vitamin D as a specific “hypercitricemic” agent.
These relationships are clinically implicated in vitamin D-deficient rickets, which results from the insufficient plasma citrate availability, particularly in infants and juveniles [1,52,57,58]. The rachitic bone is fragile and highly susceptible to fracture. The treatment with supplemental citrate and vitamin D restores the bone strength and resistance to fracture. Despite this important relationship, the mechanism by which vitamin D manifests its increased tissue citrate effect is unknown. Vitamin D induced increase in citrate production results from the inhibition of citrate oxidation, and is mimicked by fluoroacetate inhibition of citrate oxidation [56,59,60]. Fluoroacetate is a specific inhibitor of m-aconitase activity, which then prevents citrate oxidation via the Krebs cycle. Cells that conserve rather than oxidize citrate, exhibit high zinc levels, which specifically inhibits m-aconitase activity [14], and this applies to osteoblast citrate production during bone formation (Figure 2) [3,4]. It is plausible to expect that the vitamin D citrate-producing effect might involve this mechanism. Because of its important implications, the mechanism and factors involved in vitamin D induced citrate production need to be established.
Summary
• Citrate homeostasis is essential for manifestation of normal physiological activities; and its dysregulation has pathophysiological implications and clinical consequences
• The normal plasma concentration is achieved mainly by bone turnover which provides the major source of plasma citrate, and renal citrate clearance for the removal of citrate; the combination of which maintains normocitricemia
• The above is regulated by endocrine citricemic hormones in response to the plasma citrate concentration, either in combination with, or independent of, calcium regulation. Parathyroid hormone and calcitonin are respectively hypercitricemic and hypocitricemic hormones, which regulate the bone and renal activities to maintain normocitricemia
• Vitamin D is a major specific hypercitricemic agent which is achieved by promoting the activities of bone and kidney in producing and conserving citrate
• The liver does not normally remove citrate from plasma; but under specific conditions increased hepatic clearance of citrate is a major factor
• The dysregulation of plasma citrate homeostasis has pathophysiological/and clinical consequences which include: plasma calcium related defective blood clotting and impaired neuromuscular/cardiac activity: hypocitraturia and renal stone formation; bone disorders characterized by bone fragility and fractures (such as juvenile rickets and osteoporosis)
Unfortunately, the normal and dysfunctional implications of citrate homeostasis have been largely ignored by the contemporary clinical and biomedical community. The intent of this review is to bring attention to the importance of citrate homeostasis in medicine, and the need for enhanced research and funding to address the many unknown and unrecognized issues and relationships. It is in the best interest of the medical community and the public-at large that this should no longer remain a neglected area of interest and research.
Acknowledgement
The studies of LCC and RBF presented in this review were supported by NIH grants AR064808, DK076783 and CA79903.