Potentiation of ATRA Activity in HL-60 Cells by Targeting Methylation Enzymes
*Corresponding Author(s):
Ming C LiauCda Therapeutics, CA, United States
Tel:+1 8324052660,
Email:mingliau@yahoo.com
Abstract
We also evaluated Tyrosine Kinase Inhibitors (TKIs) that interfere with the production of the stabilizing factor of SAHH, and steroid analogs that compete with the endogenous steroid stabilizing factor of SAHH. While 72% of early passage (sensitive) HL-60 cells demonstrated induction of Terminal Differentiation (TD) after exposure to 1µM concentrations of ATRA, only 43% of late passage (resistant) cells showed TD. When ATRA was combined with TKI imatinib mesylate or with the steroid analogs resveratrol or β-sitosterol, agents with no innate capacity to induce differentiation, late passage HL-60 cell TD increased significantly to 98%, 99% and 94% of cells, respectively. Only modest improvements in TD percentages were seen for combinations of ATRA with cytotoxic chemotherapy agents: ATRA plus topotecan: 76%; ATRA plus oxaliplatin: 63%; ATRA plus paclitaxel: 59%. Combining ATRA with agents that interfere with maintenance of the methyl group pool potentiated its effects on an AML M2 cell line, and potentially in other forms of AML.
Keywords
INTRODUCTION
Biological generation of donor methyl groups that support DNA methylation is mediated by the ternary MMS enzyme complex consisting of MAT-MT-SAHH [6]. In the monomeric state the individual enzymes undergo rapid inactivation, while their engagement in the ternary enzyme complex promotes their stability and function. Monomeric SAHH is the most unstable, followed by MT and then MAT. Stability corresponds to their molecular size [6]. SAHH requires a stabilizing factor to assume a configuration favorable for the formation of a dimeric enzyme complex with MT, which can then form a ternary enzyme complex with MAT. In steroid hormone target tissues, such as prostate and breast, steroid hormones act to stabilize SAHH [6]. SAHH in other tissues also requires stabilizing factors similar to steroid hormones. In normal cells, SAHH is the dominant factor regulating the stability and activity of methylation enzymes. Therapeutic targeting to block stabilizing factor function can disrupt the ternary complex, leading to depletion of methyl group pools and cellular differentiation.
In cancer cells, MMS associates with telomerase (hTERT), altering the regulation and kinetic properties of the ternary enzyme complex [7,8]. Km values of the normal MAT (MATL) and hTERT-associated MAT (MATLT) are 3 µM and 20 µM methionine, respectively. Those of SAHHL and SAHHLT are 0.3 µM and 2 µM adenosine, respectively. The increased Km value of the cancer MATLT also suggests that methylation enzymes of cancer cells have elevated levels of bound S-adenosylmethionine (AdoMet), which may exercise a positive influence on the stability of ternary methylation enzymes. Binding of AdoMet by β-cystathionase has been shown to protect that enzyme against protease digestion [9]. These findings suggest that the increased Km value for MATLT in malignant cells may contribute to MMS complex stability and down-stream DNA methylation and gene silencing. Consistent with this model, it was reported that the pool size of AdoMet and S-adenosylhomocysteine (AdoHcy) was diminished in cancer cells undergoing drug-induced terminal differentiation [10]. DNA methylation maintains cell cycle transit, while incomplete methylation diverts replicating cells into terminal differentiation [11]. Therefore, factors affecting the integrity of ternary methylation enzymes are critical for cell-cycle regulation and differentiation.
ATRA, the standard therapy for APL, produces excellent initial therapeutic outcomes, with up to 90% of cases showing complete response [12]. However, remissions can be short-lived and relapse with resistance to further treatment occurs. This shortcoming, due to incomplete induction of terminal differentiation by ATRA alone, can be remedied by its use in combination therapy with drugs such as Arsenic Trioxide (ATO) [13], which is an effective DHI. Thus, a combination of DI and DHI is essential to make a perfect drug for cancer therapy.
We previously found that inhibitors of MAT and MT could potentiate ATRA induced TD of HL-60 cells at dosages not appreciably affecting the growth and differentiation of HL-60 cells [14-16]. We report here that Signal Transduction Inhibitors (STIs), polyphenols and steroids used in combination with ATRA were capable of dramatically potentiating TD of both ATRA sensitive and resistant HL-60 cells. These agents may act in part by preventing the production of, or by antagonizing stabilizing factor of SAHH, leading to decreased methyl pool generation.
MATERIALS AND METHODS
Chemicals and reagents
Culture of HL-60 cells
Nitroblue Tetrazolium (NBT) assay
Determination of potency of DHIs
RESULTS
Responsiveness of Early and Late Passage HL-60 Cells to the Induction of TD by ATRA
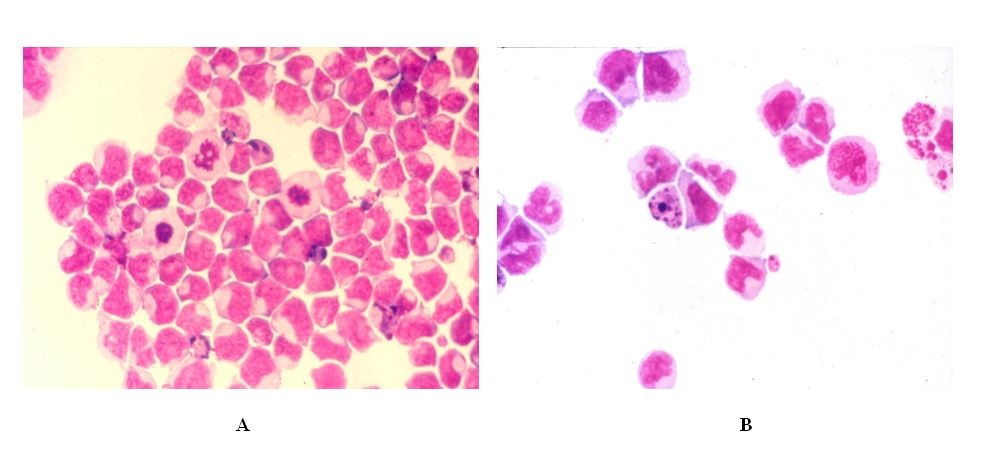 Figure 1: Hematoxylin & eosin staining of the HL60 cells before (A) and after (B) ATRA induced differentiation. The HL60 cells stained with hematoxylin (5 min) & eosin (5 min) at room temperature.
Figure 1: Hematoxylin & eosin staining of the HL60 cells before (A) and after (B) ATRA induced differentiation. The HL60 cells stained with hematoxylin (5 min) & eosin (5 min) at room temperature.|
In vitro passages |
ATRA, µm |
Final to initial cell concentration |
% Cell growth |
% NBT± |
|
|
Ratio N3/N0 |
N5/N0 |
||||
|
0-3 Months |
0 |
|
3.7 ± 0.29 |
100 |
15 ± 3.86 |
|
|
0.5 |
|
|
61 ± 2.12 |
51 ± 3.39 |
|
|
1 |
|
|
39 ± 4.36 |
72 ± 4.11 |
|
|
6 |
|
|
28 ± 3.33 |
95 ± 4.43 |
|
|
8 |
|
|
23 ± 2.12 |
88 ± 6.20 |
|
12 ± 1 Months |
0 |
4.5 ± 0.72 |
|
100 |
2 ± 1.90 |
|
|
0.5 |
|
|
72 ± 5.51 |
29 ± 2.24 |
|
|
1 |
|
|
48 ± 7.44 |
55 ± 3.91 |
|
|
6 |
|
|
40 ± 4.28 |
90 ± 5.18 |
|
|
8 |
|
|
35 ± 3.85 |
83 ± 5.58 |
|
24 ± 2 Months |
0 |
6.9 ± 1.24 |
|
100 |
0 |
|
|
0.5 |
|
|
65 ± 4.72 |
16 ± 1.95 |
|
|
1 |
|
|
54 ± 5.50 |
43 ± 3.43 |
|
|
6 |
|
|
40 ± 3.77 |
82 ± 5.62 |
|
|
8 |
|
|
35 ± 1.98 |
70 ± 4.85 |
|
36 ± 2 Months |
0 |
9.5 ± 2.72 |
|
100 |
0 |
|
|
0.5 |
|
|
79 ± 5.88 |
5 ± 2.38 |
|
|
1 |
|
|
53 ± 3.42 |
15 ± 6.21 |
|
|
6 |
|
|
47 ± 4.21 |
71 ± 5.15 |
|
|
8 |
|
|
43 ± 2.26 |
60 ± 6.34 |
|
48 ± 2 Months |
0 |
11.8 ± 3.41 |
|
100 |
0 |
|
|
0.5 |
|
|
77 ± 6.33 |
3 ± 2.12 |
|
|
1 |
|
|
55 ± 5.15 |
9 ± 4.75 |
|
|
6 |
|
|
48 ± 6.77 |
55 ± 7.48 |
|
|
8 |
|
|
40 ± 3.10 |
42 ± 3.33 |
|
60 ± 2 Months |
0 |
13.8 ± 4.28 |
|
100 |
0 |
|
|
0.5 |
|
|
83 ± 6.82 |
1 ± 0.51 |
|
|
1 |
|
|
55 ± 4.45 |
5 ± 2.27 |
|
|
6 |
|
|
47 ± 5.10 |
33 ± 4.38 |
|
|
8 |
|
|
42 ± 2.78 |
20 ± 2.63 |
Improved Induction of TD by Imatinib, resveratrol and β-sitosterol
|
Growth inhibitors |
In vitro passages of HL-60 cells |
|||
|
0-3 Months |
24 ± 2 Months |
|||
|
% Cell growth |
% NBT+ |
% Cell growth |
% NBT+ |
|
|
Imatinib mesylate, µM |
|
|
|
|
|
8 |
90 ± 5.33 |
41.5 ± 3.21 |
98 ± 4.43 |
0 |
|
16 |
78 ± 2.02 |
56.2 ± 5.66 |
90 ± 4.95 |
0 |
|
24 |
54 ± 3.55 |
63.9 ± 4.11 |
88 ± 5.05 |
0 |
|
32 |
43 ± 3.03 |
78.1 ± 6.72 |
80 ± 3.52 |
0 |
|
Resveratrol, µM |
|
|
|
|
|
0.8 |
93 ± 6.82 |
34.3 ± 3.15 |
95 ± 3.63 |
0 |
|
1.6 |
56 ± 5.66 |
55.6 ± 5.16 |
66 ± 5.35 |
0 |
|
2.4 |
37 ± 3.07 |
72.8 ± 6.22 |
49 ± 3.63 |
0 |
|
3.2 |
24 ± 2.21 |
89.1 ± 7.75 |
35 ± 2.15 |
0 |
|
β-Sitosterol, µM |
|
|
|
|
|
1.3 |
80 ± 6.30 |
29.8 ± 1.08 |
93 ± 5.25 |
0 |
|
2.6 |
48 ± 2.83 |
53.6 ± 3.82 |
78 ± 4.49 |
0 |
|
3.9 |
28 ± 1.01 |
67.8 ± 6.45 |
55 ± 2.43 |
0 |
|
5.2 |
20 ± 0.82 |
88.5 ± 7.55 |
42 ± 2.66 |
0 |
Cell incubation and NBT assay were conducted as described in table l. Imatinib mesylate, resveratrol, and β-sitosterol were dissolved in methanol. The volume applied was not to exceed 2.5%. Data are the average of at least two determinations expressed as mean ± S.D.
|
Additions |
In vitro passages of HL-60 cells |
|||
|
0-3 Months |
24 ± 2 Months |
|||
|
% Cell growth |
% NBT+ |
% Cell growth |
% NBT+ |
|
|
None |
100 |
14.8 ± i.83 |
100 |
0 |
|
1. ATRA, 0.25 µM |
85 ± 4.24 |
26.7 ± 1.40 |
|
|
|
2. ATRA, 1 µM |
|
|
54 ± 5.57 |
42.6 ± 4.35 |
|
3. Imatinib, 16 µM |
78 ± 2.12 |
56.2 ± 5.66 |
90 ± 4.95 |
0 |
|
4. Resveratrol, 1.6 µM |
56 ± 5.66 |
55.6 ± 5.16 |
66 ± 5.35 |
0 |
|
5. β-Sitosterol, 2.6 µM |
48 ± 2.83 |
53.6 ± 3.82 |
78 ± 4.49 |
0 |
|
1 + 3 |
39 ± 2.48 |
99.8 ± 0.28 |
|
|
|
2 + 3 |
|
|
44 ± 5.66 |
98.4 ± 0.64 |
|
1 + 4 |
18 ± 1.90 |
99.5 ± 0.71 |
|
|
|
2 + 4 |
|
|
14 ± 1.83 |
99.1 ± 0.85 |
|
1 + 5 |
20 ± 2.07 |
97.8 ± 2.40 |
|
|
|
2 + 5 |
|
|
22 ± 2.49 |
93.8 ± 2.35 |
Improved Induction of TD by Cytotoxic Drugs
|
Additions |
In vitro passages of HL-60 cells |
|||
|
0-3 Months |
24 ± 2 Months |
|||
|
% Cell growth |
% NBT+ |
% Cell growth |
% NBT+ |
|
|
None |
100 |
14.8 ± 1.83 |
100 |
0 |
|
1. ATRA, 0.25 µM |
85 ± 4.24 |
26.7 ± 1.40 |
|
|
|
2. ATRA, 1 µM |
|
|
54 ± 5.57 |
42.6 ± 4.35 |
|
3. Topotecan, 25 nM |
62 ± 5.13 |
35.3 ± 1.70 |
62 ± 2.83 |
0 |
|
4. Oxaliplatin, 0.63 µM |
77 ± 4.95 |
37.9 ± 0.35 |
88 ± 7.61 |
0 |
|
5. Paclitaxel, 0.25 µM |
74 ± 4.12 |
28.2 ± 0.59 |
78 ± 4.53 |
0 |
|
1+3 |
25 ± 4.24 |
80.5 ± 5.09 |
|
|
|
2+3 |
|
|
34 ± 2.31 |
76.1 ± 6.08 |
|
1+4 |
32 ± 2.12 |
66.4 ± 5.80 |
|
|
|
2+4 |
|
|
33 ± 4.24 |
62.9 ± 4.03 |
|
1+5 |
23 ± 3.07 |
55.7 ± 3.48 |
|
|
|
2+5 |
|
|
27 ± 3.34 |
58.5 ± 5.14 |
Cell culture and NBT assay were conducted as described in table 1. Topotecan and oxaliplatin were dissolved in Milli Q water and filtered through 0.2 µm membrane filters to sterilize the solutions. Paclitaxel was dissolved in methanol to apply to the medium not to exceed 2.5% of the volume. Data are the average of ≥ two determinations expressed as mean ± S.D.
Relative Potency of Growth Inhibitors or Steroid Analogs as DHIs: Dosages Needed to Achieve RI of 0.5
|
Growth inhibitors |
Dosages needed to achieve |
|
Signal transduction inhibitors |
|
|
Sunitinib malate (Sutent) |
0.28 ± 0.04 |
|
Berberine |
1.62 ± 0.17 |
|
Pazopanib (Votrient) |
10.1 ± 0.14 |
|
Imatinib mesylate (Gleevec) |
11.9 ± 2.40 |
|
Metformin |
46.9 ± 3.17 |
|
Growth inhibitors: |
|
|
As2O3 |
0.28 ± 0.11 |
|
CoCl2 |
0.62 ± 0.04 |
|
Selenite |
19.7 ± 1.56 |
|
Polyphenols |
|
|
Tannic acid |
0.37 ± 0.02 |
|
Epigallocatechin gallate |
0.62 ± 0.04 |
|
Resveratrol |
1.16 ± 0.21 |
|
Curcumin |
1.24 ± 0.16 |
|
Kuromanin |
1.43 ± 0.17 |
|
Coumestrol |
1.95 ± 0.22 |
|
Genistein |
2.16 ± 0.35 |
|
Pterostilbene |
2.19 ± 0.27 |
|
Pyrogallol |
3.18 ± 0.28 |
|
Silibinin |
3.80 ± 0.31 |
|
Caffeic acid |
3.87 ± 0.23 |
|
Ellagic acid |
4.45 ± 0.02 |
|
Gallic acid |
5.35 ± 0.12 |
|
Ferulic acid |
7.41 ± 0.15 |
|
Phloroglucinol |
38.8 ± 6.20 |
Cell culture and NBT assay were conducted as described in table 1. HL-60 cells used for the determination of reductive indices of various DHIs were maintained in vitro between 3 to 4 years. The N3/N0 ratios were between 9.5 and 11.8. Berberine, imatinib mesylate and all polyphenols were dissolved in methanol to apply to the medium not to exceed 2.5% of the volume. Sunitinib was poured out of the capsule to be extracted with HBSS in a Dounce homogenizer. Pazopanib and metformin was extracted with HBSS in a Dounce homogenizer. Insoluble materials were removed by centrifugation at 1200xg for 10 min. CoCl2 was dissolved in HBSS. As2O3 and selenite was suspended in HBSS with phenol red indicator. 1 N NaOH was added drop wise until pink color persisted. All aqueous preparations were sterilized by passing through 0.2 µm membrane filters. Reductive indices were obtained as described in Materials and Methods. Dosages of DHIs needed to achieve reductive index of 0.5 were obtained from the plots of reductive indices versus concentrations. Data are the average of ≥ two determinations expressed as mean ± S.D.
|
Steroid analogs |
Dosages needed to achieve |
|
reductive index of 0.5, µM |
|
|
Vitamin D3 |
0.61 ± 0.11 |
|
Dexamethasone |
0.75 ± 0.20 |
|
Testosterone |
1.55 ± 0.13 |
|
Gugulsterone |
1.59 ± 0.16 |
|
β-Sitosterol |
1.72 ± 0.02 |
|
Dehydroepiandrosterone |
1.79 ± 0.24 |
|
Dihydrotestosterone |
2.10 ± 0.20 |
|
Prenisolone |
2.22 ± 0.12 |
|
Estradiol |
2.45 ± 0.02 |
|
Progesterone |
3.55 ± 0.18 |
|
Hydrocortisone |
4.59 ± 0.23 |
|
Pregnenolone |
7.16 ± 1.13 |
|
Pregnenolone sulfate |
7.35 ± 1.06 |
Cell culture, NBT assay, and determination of reductive indices were conducted as described in table 5. Data are the average of ≥ two determinations expressed as mean ± S.D.
STIs are in general small molecule inhibitors of protein tyrosine kinases. Aberrant activation of tyrosine kinases secondary to mutations produce excess growth signals that result in the production of stabilizing factors for SAHH. We tested a variety of TKIs with ATRA, including sunitinib malate, a multikinase inhibitor, berberine, a potent inhibitor of EGFR-MEK-ERK signaling pathway, pazopanib, a potent inhibitor of the vascular endothelial growth factor, and imatinib mesylate, which targets Bcr-Abl in Philadelphia chromosome positive chronic myeloid leukemia and CD117 (cKIT) in gastrointestinal stromal tumors [22-25]. We also evaluated metformin, a well-known oral hypoglycemic agent, in combination with ATRTA based on its capacity to inhibit mTOR [26,27]. Among TKIs studied, sunitinib malate had the most potent activity as a DHI. More selective TKIs, such as pazopanib and imatinib mesylate, were less active (Table 5). Broad spectrum TKIs may be better candidates as DHIs.
Among non-specific growth inhibitors studied, ATO and CoCl2 had impressive activity as DHIs (Table 5). ATO, although quite toxic, required very low dosages to function as a DHI. CoCl2, an agent that up regulates hypoxia inducible factor, induced HL-60cell attachment. When cells became attached to the culture dish, cell growth was greatly diminished. The induction of cell attachment may enable CoCl2 to act as a DHI. Sodium selenite is linked to an array of health benefits, including prevention of cancer. However, its cancer fighting potential has never been well characterized. Its activity as a DHI was limited. As listed in table 5, we found that several polyphenols had impressive activity as DHI’s. Many of these polyphenols are present in foods that are regularly consumed. The activity of polyphenols may be attributable to inhibition of signaling pathways [18, 23,28-31].
Steroid Analogs as Differentiation Helper Inducers
DISCUSSION
Our study showed dramatic improvement in the induction of TD after combined therapy with ATRA with TKIs or steroid analogs. This is thought to occur as a result of blockade of growth signals and methyl group transfer associated with gene silencing (Figure 2). These approaches may offer clinical utility for the treatment of ATRA-resistant clones. As shown in table 1, HL-60 cells gradually became resistant to ATRA-induced TD during prolonged propagation in vitro. Gene silencing secondary to abnormal methylation enzyme activity is thought to play a role in the development of ATRA resistance in AML [2]. Our data suggest that a multi agent approach combining agents that disrupt DNMT activity in conjunction with targeting RARA can produce favorable therapeutic results. The use of ATRA alone could stimulate at most 48% of resistant HL-60 cells to undergo TD, whereas ATRA in combination with DHI agents led to TD in almost 100% of the resistant cells.
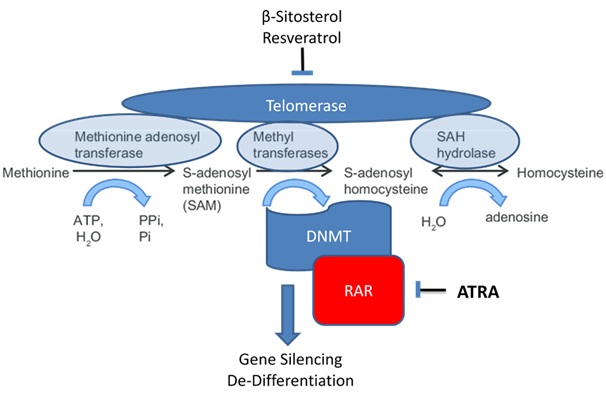 Figure 2: Mechanism of action of ATRA in combination with agents that disrupt the telomerase/methyltransferase complex.
Figure 2: Mechanism of action of ATRA in combination with agents that disrupt the telomerase/methyltransferase complex.Hypoxic growth in vivo is another element that could contribute to ATRA resistance. It has been shown that cancer stem cells able to undergo differentiation in normoxia were unable to undergo differentiation in hypoxia [36,37]. Hypoxia influences the switch between differentiation and stemness [36]. The enhanced expression of telomerase in hypoxia may be responsible for the blockade of differentiation. Cancer stem cells already express an unusually high level of telomerase [38], which has to be down-regulated for differentiation to take place [39]. Hypoxia Inducible Factor (HIF), stabilized under hypoxic conditions, is a transcription cofactor that up regulates telomerase, leading to stabilization of the MMS complex [40]. Resveratrol and topotecan treatment lead to decreased HIF under hypoxic conditions [41,42]. Consequently, resveratrol and topotecan may be candidates for targeting cancer stem cells. They were both found to promote TD in late passage HL-60 cells. Taken together, these data support the notion that ATRA in combination with DHI’s may be of value in the treatment of non-M3AML.
CONFLICT OF INTEREST
ACKNOWLEDGEMENT
REFERENCES
- Lyko F (2018) The DNA methyltransferase family: a versatile toolkit for epigenetic regulation. Nat Rev Genet 19: 81-92.
- Figueroa ME, Lugthart S, Li Y, Erpelinck-Verschueren C, Deng X, et al. (2010) DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17: 13-27.
- Liau MC, Hunt ME, Hurlbert RB (1976) Role of ribosomal RNA methylases in the regulation of ribosome production. Biochemistry 15: 3158-3164.
- Bernstein KA, Bleichert F, Beach JM, Cross FR, Baserga SJ (2007) Ribosome Biogenesis is Sensed at the Start Cell Cycle Checkpoint. Mol Cell Biol 18: 953-964.
- Jones PA, Issa JP, Baylin S (2016) Targeting the cancer epigenome for therapy. Nat Rev Genet 17: 630-641.
- Liau MC, Chang CF, Saunders GF, Tsai YH (1981) S-Adenosylhomocysteine hydrolases as the primary target enzymes in androgen regulation of methylation complexes. Arch Biochem Biophys 208: 261-272.
- Liau MC, Zhu PZ, Chiou GCY (2010) Identification of the tumor factor of abnormal cancer methylation enzymes as the catalytic subunit of telomerase. Clinical Oncology and Cancer Research 7: 86-96.
- Liau MC, Chang CF, Becker FF (1979) Alteration of S-adenosylmethionine synthetase during chemical hepatocarcinogenesis and in resulting carcinomas. Cancer Res 39: 2113-2119.
- Prudova A, Baumann Z, Braun A, Vitvitsky V, Lu SC, et al. (2006) S-Adenosylmethionine stabilizes β-cystathionine synthase and modulates redox capacity. Proc Natl Acad Sci U S A 103: 6489-6494.
- Chiba P, Wallner L, Kaizer E (1988) S-Adenosylmethionine metabolism in HL-60 cells: effect of cell cycle and differentiation. Biochim Biophy Acta 971: 38-45.
- Liau MC, Lee SS, Burzynski SR (1989) Hypomethylation of nucleic acids: a key to the induction of terminal differentiation. Intl J Exp Clin Chemother 2: 187-199.
- Hu J, Liu YF, Wu CF, Xu F, Shen ZX, et al (2009) Long term therapy and safety of all-trans retinoid acid/arsenic trioxide-based therapy in newly diagnosed acute promyelocytic leukemia. Proc Natl Acad Sci USA 106: 3342-3347.
- Degos L, Wang ZY (2001) All trans retinoic acid in acute promyelocytic leukemia. Oncogene 20: 7140-7145.
- Liau MC, Liau CP, Burzynski SR (1992) Potentiation of induced terminal differentiation by phenylacetic acid and related chemicals. Int J Exp Clin Chemother 5: 9-17.
- Liau MC, Huang LJ, Lee JH, Chen SC (1998) Development of differentiation helper inducers for differentiation therapy of cancer. Chin Pharm J 50: 289-303.
- Liau MC, Liau CP (2002) Methyltransferase inhibitors as excellent differentiation helper inducers for differentiation therapy of cancer. Bull Chin Cancer 11: 166-168.
- Blair OC, Carbone R, Sartorelli AC (1985) Differentiation of HL-60 promyelocytic leukemia cells monitored by flow cytometric measurement of nitro blue tetrazolium (NBT) reduction. Cytometry 6: 54-61.
- Liu M, Wilk SA, Wang A, Zhou L, Wang RH, et al. (2010) Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem 285: 36387-36394.
- Kundu JK, Surth YJ (2008) Cancer chemoprevention and therapeutic potential of resveratrol: mechanism perspectives. Cancer Lett 269: 243-261.
- Ju YH, Clausen LM, Allred KF, Almada AL, Helferich WG (2004) beta-Sitosterol, beta-Sitosterol Glucoside, and a Mixture of beta-Sitosterol and beta-Sitosterol Glucoside Modulate the Growth of Estrogen-Responsive Breast Cancer Cells In Vitro and in Ovariectomized Athymic Mice. J Nutr 134: 1145-1151.
- Liau MC, Lee SS, Burzynski SR (1990) Modulation of cancer methylation complex isozymes as a decisive factor in the induction of terminal differentiation mediated by Antineoplaston A5. Intl J Tiss React 12: 27-36.
- Demetri GD, van Oosterom AT, Garrett CR, Blackstein ME, Shah MH, et al. (2006) Efficacy and safety of sunitinib in patient with advanced gastrointestinal stroma tumor after failure of imatinib: a randomized controlled trial. Lancet 368: 1329-1338.
- Liu G, Xu X, Zhao M, Wei Z, Li X, et al. (2015) Berberine induces senescence of human glioblastoma cells by down regulating the EGFR-MEK-ERK signaling pathway. Mol Cancer Ther 14: 355-363.
- Harris PA, Boloor A, Cheung M, Kumar R, Crosby RM, et al. (2008) Discovery of 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem 15: 4632-4640.
- DiNitto JP, Wu JC (2011) Molecular mechanisms of drug resistance in tyrosine kinases cAbl and cKit. Critical Reviews in Biochemistry and Molecular Biology 46: 295-309.
- Sahra IB, Regazzeti C, Robert G, Laurent K, Marchand-Brustel YL, et al. (2011) Metformin, independent of AMPK, induces mTOR inhibition and cell cycle arrest through REDD1. Cancer Res 71: 4366-4372.
- Rocha GZ, Dias MM, Ropelle ER, Osório-Costa F, Rossato FA, et al. (2011) Metformin amplifies chemotherapy induced AMPK activation and antitumor growth. Clin Cancer Res 17: 3993-4005.
- Liao S, Xia J, Chen Z, Zhang S, Ahmad A, et al. (2011) Inhibitory effect of curcumin on oral carcinoma CAL-27 cells via suppression of Notch-1 and NF-kB signaling pathways. J Cell Biochem 112: 1055-1065.
- Moon SS, Kim MO, Xhoi YH, Lee HG, Kim ND, et al. (2008) Gossypol suppresses telomerase activity in human leukemia cells via regulation hTERT. FEBS Lett 582: 3367-3373.
- Yang G, Fu Y, Malakhova M, Kurinov I, Zhu F, et al. (2014) Caffeic acid directly targets ERK1/2 to attenuate solar UV-induced skin carcinogenesis. Cancer Prev Res 7: 1056-1066.
- Kim PK, Suh Y, Yoo KC, Cui YH, Hwang E, et al. (2015) Phloroglucinol suppresses metastatic ability of breast cancer cells by inhibition of epithelial-messenchymal cell transition. Cancer Sci 106: 94-101.
- van Gils N, Verhagen HJMP, Smit L (2017) Reprogramming acute myeloid leukemia into sensitivity for retinoic-acid-driven differentiation. Exp Hematol 52: 12-23.
- Phi LTH, Sari IN, Yang YG, Lee SH, Jun N, et al. (2018) Cancer stem cells (CSCs) in drug resistance and their therapeutic implications in cancer treatment. Stem Cells International 2018: 1-16.
- McCracken MN, George BM, Kao KS, Marjon KD, Raveh T, et al. (2016) Normal and Neoplastic Stem Cells. Cold Spring Harb Symp Quant Biol 81: 1-9.
- Liau MC (2004) Abnormal methylation enzymes: a selective molecular target for differentiation therapy of cancer. Chin Pharm J 56: 57-67.
- Fruehauf JP, Liau MC (2011) Therapeutic targeting of HIF to reverse the cancer stem cell phenotype and epithelial-mesenchymal transition. Therapy 8: 737-740.
- Kung AL, Zablludoff SD, Fraces DS, Freedman SJ, Tanner EA, et al. (2004) Small molecule blockade of transcriptional activation of hypoxia inducible factor pathway. Cancer Cell 6: 33-43.
- Shay JN, Wright NE (2010) Telomeres and telomerase in normal and cancer stem cells. FEBS Lett 584: 3819-3825.
- Reichman TW, Albanell J, Wang X, Moore MA, Studzinski GP (1997) Downregulation of telomerase activity in HL60 cells by differentiating agents is accompanied by increased expression of telomerase-associated protein. J Cell Biochem 67: 13-23.
- Anderson CJ, Hoare SF, Ashcroft M, Bilsland AE, Keith WN (2006) Hypoxic regulation of telomerase gene expression by transcriptional and post-transcriptional mechanisms. Oncogene 25: 61-69.
- Sun Y, Wang H, Liu M, Lin F, Hua J (2015) Resveratrol abrogates the effects of hypoxia on cell proliferation, invasion and EMT in osteosarcoma cells through downregulation of the HIF-1α protein. Mol Med Rep 11: 1975-1981.
- Rapisarda A, Shoemaker RH, Melillo G (2004) Targeting topoisomerase I to inhibit hypoxia inducible factor 1. Cell Cycle 3: 172-175.
Citation: Liau MC, Kim JH, Fruehauf JP (2019) Potentiation of ATRA Activity in HL-60 Cells by Targeting Methylation Enzymes. J Pharmacol Pharmaceut Pharmacovig 3: 009.
Copyright: © 2019 Ming C Liau, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.