A Model to Study the Inhibition of Arginase II with Noscapine&Its Derivatives
*Corresponding Author(s):
Prashant SinghDepartment Of Chemistry, Atma Ram Sanatan Dharma (ARSD) College, University Of Delhi, New Delhi, India
Tel:+911124113436,
Email:psingh@arsd.du.ac.in
Abstract
Background and Purpose
Nitrate tolerance can be explained based on the reduction of the vessel responsiveness and the same is used for endogenous vasodilator Nitric Oxide (NO). There are some limitations for the treatment of ischaemia, angina etc. and it attracted the scientists and researchers. The location of arginase II is endothelial cells in mitochondria and it is used to change the potency of endothelial nitric oxide synthase.
Experimental approach
A theoretical model has been developed to find the potent arginase II inhibitor. A library of noscapine (116 molecules) was designed and optimized using computational tools. Then, the designed molecules were docked with the arginase II (PDB: 4IXU) using iGemdock. Based on binding energy, the potential candidate was screened. Further, Absorption Distribution Metabolism, Excretion and Toxicity (ADMET) using online web-server and Density Functional Theory (DFT) study of the top four screened molecules has been studied by using Gaussian. Then, molecular dynamic simulation of arginase II with and without 97 was performed using Gromacs. Further, the binding energy was determined using MM-PBSA on Gromacs.
Conclusion
Compound no. 97 showed the best binding with the arginase II based on docking. Further, the potency of the screened noscapine 97against arginase II was compared with the reported molecules. MD simulations showed the stable anchoring of the arginase II-97 complex and the binding energy between 97 and arginase II was found to be negative i.e. -815.184 kcal/mol.
Keywords
Density functional theory; MM-PBSA; Molecular dynamics simulation, Noscapine; Protein data bank.
INTRODUCTION
Angina pectoris is an uncomfortable condition, like chest pain and it is due to less oxygen supply to the coronary artery [1,2]. Endothelium has an important role in deciding the role in the release of Endothelial-Derived Relaxing Factor (EDRF) [3]. The major compound Nitric Oxide (NO) regulates the arterial pressure by dilating the blood vessels [4]. NO is synthesized from arginine by means of endothelial Nitric Oxide Synthase (eNOS) [5]. Many research groups have focused to develop the bioactive compounds to alter the L-arginine metabolism in the body.
Arginase-II catalyzes the degradation of arginine into ornithine and urea [6]. Arginase-IIis responsible for the bioavailability of L-arginine for Nitric Oxide Synthase (NOS) by the mean of the competition of substrate [7]. Therefore, when the activity of arginase is increased, it causes diseases by reducing the amount of L-arginine in the body. It is needed by NOS to produce NOe [6-8]. In the last few decades, researchers showed great interest in studying the role of arginase in the cure of diseases. Various arginase inhibitors have been reported and have shown potential under different pathophysiological conditions like renal injury in diabetic [9], atherosclerosis [10], erectile dysfunction and pulmonary hypertension [11,12], hypertension [13], allergic rhinitis [14] and many more.
Phthalideisoquinilines based alkaloids are popular molecules and cones under the class of isoquinoline based compounds viz. erythro and threo form. Noscapine containsisoquinoline and benzofuran ring as an active ingredient. Primarily it is used as an antitussive agent to suppressa cough. At present, noscapine its its derivatives are explored and under clinical trials for the treatment of different diseases like cancer [15,16]. There is too much structural variability in noscapine, which make its use for different purposes.
There is need to find the arginase II inhibitor to control or cure angina. The potential of the erythro form of noscapine against the arginase-II has been investigated. In the present work, a theoretical model for the inhibition of arginase-II by noscapine and its derivatives was developed. Molecular docking, density functional theory, Absorption Distribution Metabolism, Excretion and Toxicity (ADMET), Molecular Dynamic (MD) simulations along with Molecular Mechanics Poisson–Boltzmann Surface Area (MM-PBSA) analysis were performed to find the potent arginage II inhibitor.
EXPERIMENTAL
This experimental work is categorized into five parts i.e. designing of molecules &molecular docking, ADMET studies, DFT studies, MD simulations along with MM-PBSA analysis. The overall experimental approach of the work can be understood by flowchart 1.
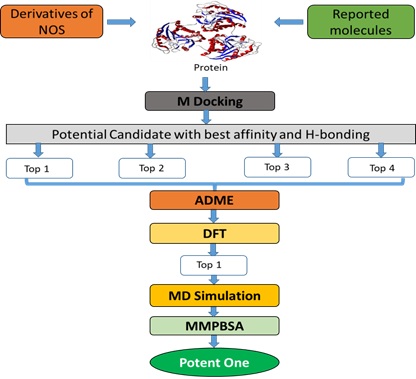 Flowchart 1: The overall methodologies used in the whole work.
Flowchart 1: The overall methodologies used in the whole work.
Designing of molecules and molecular docking
Designing of molecules
There are two isomeric form of noscapine viz., erythro and threo form. Herein, only erythro-form of noscapine was considered due to its high stability and biological potential. In the present work, a total of116 molecules based on noscapine were designed as in table1.
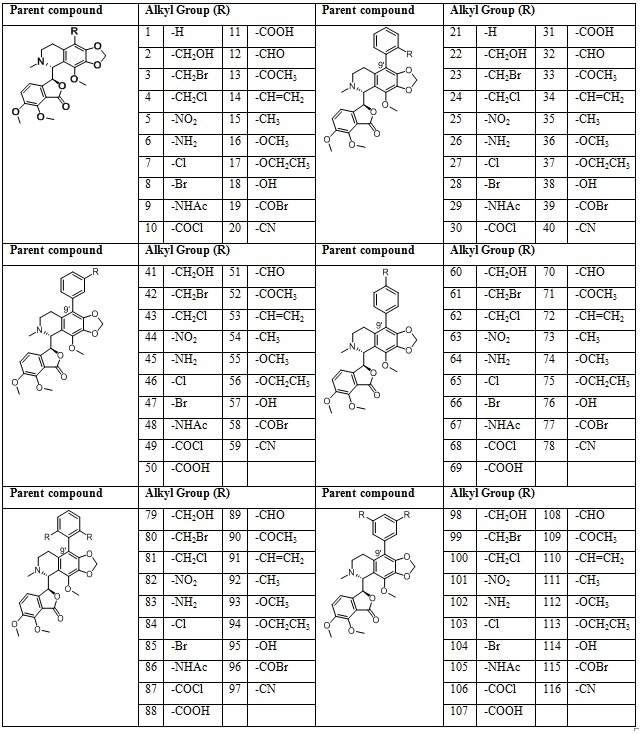 Table 1: Libraries of the noscapine derivatives
Table 1: Libraries of the noscapine derivatives
15 reported arginase II inhibitor were also taken from the literature for a comparison with noscapine as in table 2.
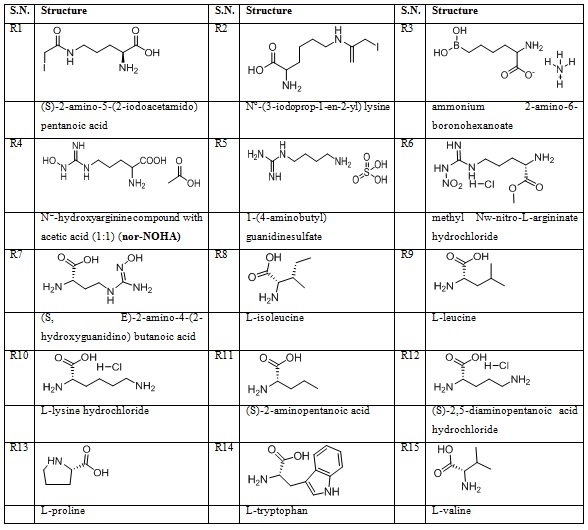
Table 2: 15 reported arginase II inhibitors.
Geometry optimization of noscapines & reported molecules
The designing of all compounds was done by using ACD Che sketch and their optimization was done by choosing molecular mechanics (MM2) as a force field. These optimized compounds were used for docking.
Protein preparation
Protein preparation was done by using Molegro Molecular Viewer (MMV 2.5). The following parameters were checked like flexible torsion in compounds, missing charges, assigning of bonds, tripos type atoms and missing explicit hydrogen. The prepared protein was used for the docking analysis and MD simulation.
Molecular Docking
The docking of all noscapines (Table 1) and the reported molecules (Table 2) was performed using iGemdock [17] against the arginase-II (PDB ID - 4IXV). This software used the generic algorithms for the docking.
Docking parameters & Post Docking modeling
Herein, the parametersfor the docking are set with population size of 200 and generation of 70 along with two solutions for each. On the basis of the above set parameters, the compounds were screened [17]. The top four compoundswereselected by considering the lowest binding energy, can be determined by the equation 1.
EBinding = EVDW + Hbond + Elec (1)
VDW - vander Waal energy;
Hbond- hydrogen bonding energy;
Elec - electro statistic energy
The modelling of the docked poses is studied by Discovery studio visualizer v 3.5 [18].
ADMET properties
The ADME (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties were calculated to check the better bioavailability of the proposed drug molecule.
Physicochemical parameters
The physiochemical properties like Log S, Solubility, number of heavy atoms, number of rotatable bonds, number H-bond acceptors, number H-bond donors, Log Po/w, and physiochemical space for oral availability were checked by the web server (http://www.swissadme.ch/) [19].
Biological properties
The biological properties like TPSA (Ų), GI absorption, BBB permeant, P-gp substrate, and CYP3A4 inhibitor were calculated by the web server (http://www.swissadme.ch/). Absorption (% ABS) of top four molecules was calculated according to the method described by Zhao et al. [20] TPSA is an important factor to give an idea for ability of drug transport and can be determined by using equation 2. The results are incorporated in table 4.
%ABS = 109 – [0.345 × topological polar surface area (TPSA) (2)
Other biological properties like GPCR ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor, andenzyme inhibitor value by using the online server molinspiration (www.molinspiration.com) [21].
Toxicity
The acute rat toxicity of the top four moleculeswas calculated using an online server GUSAR (http://www.way2drug.com/gusar/acutoxpredict.html). The toxicity parameters like IP LD50, IV LD50, Oral LD50, and SC LD50 for all four routes of administration i.e., oral, intraperitoneal, intravenous, and subcutaneous for top four molecules were calculated. This toxicity model was based on a rat [22].
DFT analysis
Density Functional Theory (DFT) have been performed to study the electrical properties of the noscapine derivative. Geometry optimization of the molecules were performed. Becke’s 3 parameters functional Lee, Yang, Parr B3LYP/6-311++G (d, p) was used for the calculation with the Gaussian 09 [23]
In addition, DFT is very useful in providing chemical descriptors such as chemical hardness (η), chemical potential ( ), electronegativity (χ), softness (S), and global electrophilicity index (ω), these are given in equation 3-7[24].
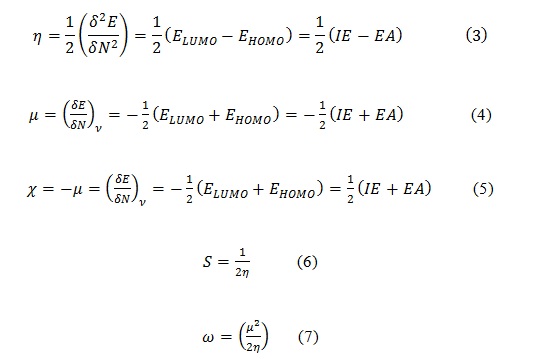
Where IE is ionization potential and EA is electron affinity.
MD Simulation
The Molecular Dynamics (MD) is a technique used to study the fundamental structural response of protein with and without ligand at the nanoscale [25]. MD analysis of protein and protein-ligand complex was done by using GROMACS 5.1.4 [26]. CHARM force-field parameters was used in analysis [27]. The topology and coordinates of the ligandwere generated by Swiss Param online web server [28]. The system was solvated by water in the cubic box manner taking simple point charge model to develop a Periodic Boundary Condition (PBC). Na+ and Cl- ions were used to neutralise the system. The energy minimization of the system was performed by applying a steepest descent algorithm with 1000 steps to release the conflict contacts.
MD simulationsanalysis were performed in two phases (i) ensemble equilibration and (ii) MD production. The temperature and pressure equilibration were performed to control the temperature and pressure of the system, the system is warmed to 300 K at 1 bar for 100 ps using leap-frog integrator for temperature coupled by modified Berendsen thermostatand leap-frog integrator for pressure coupled by Parrinello-Rahman. MD simulations were done taking cut off the size of 12 Å. Particle Mesh Ewald (PME) method was used for all long-range electrostatics’ charges. The MD production is run for 10ns and the coordinates were recorded on an interval of 10 ps.
MM-PBSA analysis
MM-PBSA analysis is done after the screening and done by using the trajectories obtained from the MD simulation. MM-PBSA study for the complex between the ligand and arginase-II was studied by the g_mmpbsa [29-30]. This method provides the change in free energy for the formation of complex. Various binding free energy change were calculated for the complex with the help of the equations 8-15 respectively [29-30].

The ensemble hypothesis was used to calculate these all changes from the coordinates of corrected MD trajectories [29].
RESULTS AND DISCUSSION
Molecular Docking
Dockingis a computational methodology to study the binding of small molecules with a receptor to forma complex to enhance or inhibit the biological potency of protein [31]. The total binding energy of all docked molecules are used to screen andthe top four molecules i.e., ligand 97, 109, 48 and 101 are mentioned in table 3 along with the 15 reported inhibitors. Ligand 97shows the highest negative value against 4IXV, which is -133.413KJ/mol. The designed top four molecules shows strong binding energy than the reported molecules. The binding energy values for ligand 109, 48 and 101 are -129.175, -126.677 and -126.298 respectively. The highest value in reference molecule is for the 1-(4-aminobutyl) guanidinesulfate (R5), its value is -110.052. Table3 shows that noscapines can be used to inhibit the function of arginase-II more effectively than the reference molecules. The docking results of he designed noscapines are available in supporting file. Docking data of all the designed noscapines are given in Supplementary information as in table Sa.
|
Ligand |
EBinding |
EVDW |
EH-bonding |
EElect |
|
Noscapine Derivatives |
||||
|
97 |
-133.413 |
-93.6378 |
-39.7757 |
0 |
|
109 |
-129.175 |
-115.069 |
-14.1058 |
0 |
|
48 |
-126.677 |
-98.9796 |
-27.6971 |
0 |
|
101 |
-126.298 |
-95.8055 |
-28.7223 |
-1.7701 |
|
Reference Molecule |
||||
|
R5 |
-110.052 |
-54.3164 |
-55.1636 |
-0.57243 |
|
R4 |
-92.2519 |
-50.6993 |
-42.7032 |
1.15061 |
|
R7 |
-87.7108 |
-46.2689 |
-39.0585 |
-2.38333 |
|
R3 |
-87.0116 |
-53.3087 |
-35.0249 |
1.32208 |
|
R6 |
-84.8008 |
-56.2746 |
-28.5261 |
0 |
|
R14 |
-79.4946 |
-54.3613 |
-20.653 |
-4.48028 |
|
R2 |
-73.7685 |
-46.8754 |
-27.4161 |
0.522978 |
|
R1 |
-72.7108 |
-45.508 |
-22.9159 |
-4.28693 |
|
R12 |
-67.9846 |
-50.7546 |
-17.23 |
0 |
|
R11 |
-64.3878 |
-34.752 |
-32.4239 |
2.78805 |
|
R8 |
-64.1126 |
-42.7747 |
-27.2768 |
5.93892 |
|
R10 |
-63.2022 |
-40.3005 |
-20.1627 |
-2.73904 |
|
R13 |
-63.1236 |
-46.367 |
-23.5179 |
6.76133 |
|
R9 |
-62.9151 |
-46.7523 |
-22.8751 |
6.71231 |
|
R15 |
-62.1513 |
-35.458 |
-29.7164 |
3.02315 |
Table 3: Docking score of the top four noscapine derivatives and reported molecules
The configurational analysis of the active site of a protein is also done to check the active amino acid residues which bind with the noscapines. It is found that the noscapines targeted same active site of arginase II occupied
by the first-generation arginase inhibitors like NOHA. It is clearly understood that noscapines can be used to inhibit arginase-II activity more effectively than the reported inhibitors. Ligand 97 shows H-bond interaction given in figure 1a with SER-156 (2.33 Å) and ASN-158 (2.33695 Å & 1.93419 Å), ligand 109 shows H-bond interaction given in figure 1b with GLU-275 (2.57936 Å) and with TYR-273 (2.35556Å), ligand 48 shows H-bonds interaction given in figure 1c with THR-310 (2.81668 Å), ARG-57 (2.08954 Å), GLN-325 (1.92582 Å) and ASP-317 (2.48569 Å) with and ligand 101 don’t show any H-bond interaction given in figure 1d.
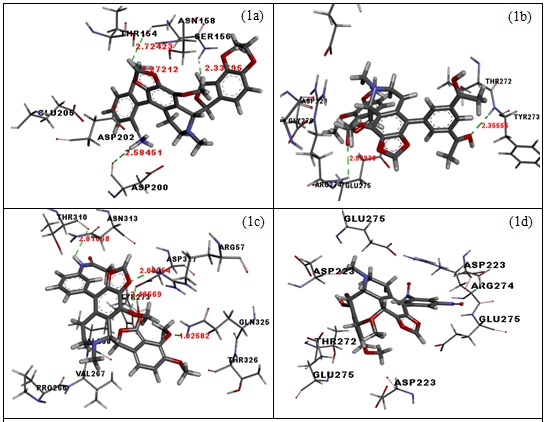 Figure 1(a-d): H-bond poses of 97, 109, 48 and 101 with amino acid of arginase-II.
Figure 1(a-d): H-bond poses of 97, 109, 48 and 101 with amino acid of arginase-II.
The contribution of amino acids of binding pockets within and around 8 Å of the ligand was also analyzed and a graph of amino acid versus total binding energy is also plotted for the ligand 97, 109, 48 and 101 (Graph 1 a-d). The major amino acid contribution of active cavity for ligand 97 are ASN-147, SER-155, ASN-158, ASP-200, ASP-202, GLY-161, GLU-205 and THR-265, for ligand 109 are TYR-273, THR-272, GLU-275, ASP-256, ARG-274, GLY-227, GLY-269 and ASP-223, for ligand 48 are ARG-57, TYR-273, THR-310, ASP-317, GLN-325, ASN-313, ARG-119, ASP-200, VAL-267 and VAL-268 and for ligand 101 are ASP-223, ARG-274, ILE-227, THR-272, GLU-275 and GLY-226. From the binding cavity residues analysis, it is clear that the main residues which are the part active cavity are mainly composed of ASN-147, SER-155, ASN-158, ASP-200, ASP-202, GLY-161, GLU-205, THR-265, TYR-273, THR-272, GLU-275, ASP-256, ARG-274, GLY-227, GLY-269, ASP-223, THR-310, ASP-317, GLN-325, ASN-313, VAL-267, VAL-268,andILE-227.
 Graph 1(a-d): Showing cavity residues and their negative contribution in the stabilization of Ligand 97, 109, 48 and 101 respectively.
Graph 1(a-d): Showing cavity residues and their negative contribution in the stabilization of Ligand 97, 109, 48 and 101 respectively.
In Graph 2, the structural properties of the arginase II 4IXV was determined using the SAVES server (online) and the analysis was explained as in Graph 2 regarding the allowed and disallowed regions.
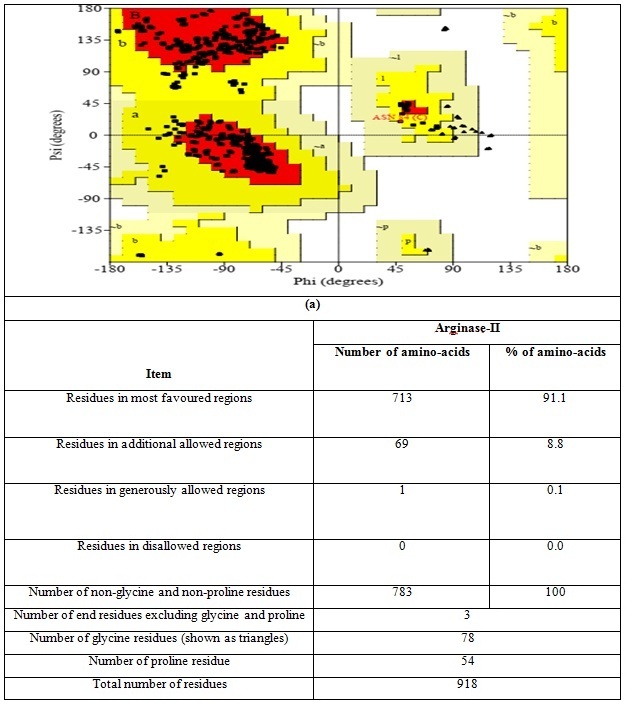
Graph 2 (a) the ramachandran plot and (b) the 3D structures analysis of Arginase-II
ADMET analysis
ADMET properties of the molecules are generally more useful when a new molecule is proposed as a potential drug [32]. Oral bioavailability of a drug can be explained as a part of oral drug in systematic circulation in the body. Oral absorption depends on the permeability and aqueous solubility of a molecule. It can be controlled by molecular weight, logP, Topological Polar Surface Area (TPSA), no. of rotatable bonds, log S, number of Hydrogen Bond Donors (HBD) and Acceptors (HBA) [33]. It was found that noscapine followed the Lipinski’s “rule of five” with one violation. Although, the aqueous solubility of noscapinewas found to be low and should need to increase e (Table 4). For four ligands 97, 109, 48 and 101the log P value are 3.9, 4, 4 and 3.18 respectively, which is under 5, the log S values are -5.56, -5.58, -5.31 and -5.80 respectively and are moderately soluble. The maximum no. of allowed rotatable bonds should be 9, and all top four moleculesare under the allowed limit. The allowed limit of no. of H-bond acceptor is 10 and donor should be 5, and here ligand 97, 109, and 48 are under the allowed limit. The physiochemical oral availability was also calculated to top four ligands and it is found that the top three under the space. ADME properties of all the designed molecules is given in Supplementary information as table Sb.
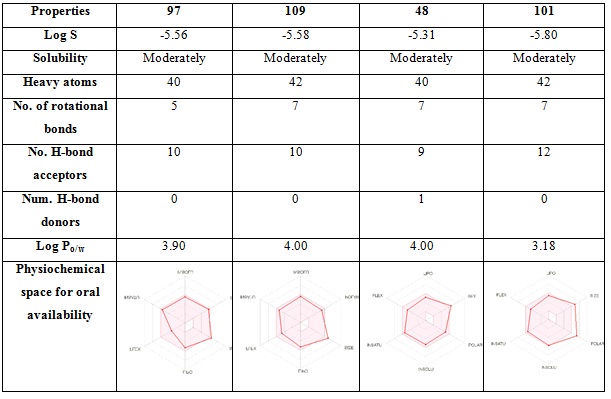
Table 4: Physiochemical descriptors of top four noscapine derivatives
Biological properties like % absorbance from TPSA, Gastro Intestinal (GI) absorbance, Blood-brain Barrier (BBB) permeation, permeability glycoprotein (P-gp) substrate value, cytochrome P450 (CYP3A4) inhibition value, molinspiration Log P (miLog P), Globular Protein-coupled Receptor (GPCR) inhibition value, ion channel modulator, kinase inhibitor, nuclear receptor, protease inhibitor, and enzyme inhibition value were calculated and are given in table 5. The GI absorption value for top three ligands is found to high while for ligand 101 is low. The BBB value for all top four molecules is found negative, the P-gp is found as positive, cytochrome P450 (CYP3A4) inhibition value for top three positives, miLog P is below 5, the GPCR value is found positive for ligand 97 and 48, and for 109 and 101 is negative, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor, and enzyme inhibitor value is negative for all, while the lowest score is for ligand 97 among top four, which shows the ligand 97 has more biological potential among the top four.
|
Properties |
97 |
109 |
48 |
101 |
|
TPSA (Ų) |
123.27 Ų |
109.83 Ų |
104.79 Ų |
167.33 Ų |
|
%ABS |
66.47185 |
71.10865 |
72.84745 |
51.27115 |
|
GI absorption |
High |
High |
High |
Low |
|
BBB permeant |
No |
No |
No |
No |
|
P-gp substrate |
Yes |
Yes |
Yes |
Yes |
|
CYP3A4 inhibitor |
Yes |
Yes |
Yes |
No |
|
miLogK |
3.95 |
4.26 |
3.73 |
4.38 |
|
GPCR ligand |
0.07 |
-0.01 |
0.02 |
-0.09 |
|
Ion channel modulator |
-0.13 |
-0.37 |
-0.24 |
-0.38 |
|
Kinase inhibitor |
-0.26 |
-0.50 |
-0.34 |
-0.49 |
|
Nuclear receptor ligand |
-0.30 |
-0.46 |
-0.45 |
-0.52 |
|
Protease inhibitor |
-0.31 |
-0.34 |
-0.35 |
-0.40 |
|
Enzyme inhibitor |
-0.03 |
-0.20 |
-0.13 |
-0.24 |
Table 5: Biological of the top hit four molecules against arginase-II
The toxicity estimation of compounds wastested as QSAR model based on the rat toxicity, where the Lethal Dose (LD) 50 were calculated for the fours of administration i.e. intraperitoneal, intravenous, oral and subcutaneous (Table 6). The ligand 97, 109, 48 and 101 all came in applicability domain of model while the subcutaneous LD50 value for the ligand 97 falls out of applicability domain. All the LD50 value for the ligand 97 is almost the lowest in all top four ligands.
|
Properties |
97 |
109 |
48 |
101 |
|
Rat IP LD50 (mg/kg) |
577,400 in AD |
605,000 in AD |
528,800 in AD |
419,200 in AD |
|
Rat IV LD50 (mg/kg) |
30,580 in AD |
36,110 in AD |
31,610 in AD |
33,730 in AD |
|
Rat Oral LD50 (mg/kg) |
947,900 in AD |
1576,000 in AD |
1202,000 in AD |
885,300 in AD |
|
Rat SC LD50 (mg/kg) |
1256,000 out of AD |
782,300 in AD |
645,000 in AD |
468,400 in AD |
|
Note: |
IP - Intraperitoneal route of administration |
IV - Intravenous route of administration |
Oral - Oral route of administration |
SC - Subcutaneous route of administration |
|
in AD - compound falls in applicability domain of models |
out of AD - compound is out of the applicability domain of models |
|||
Table 6: Toxicity of the top hit four molecules against arginase-II
DFT Analysis
DFT studies help to understand molecular properties and the behavior of atoms in molecules. Hard molecules have a large HOMO-LUMO gap and soft molecules showed a reverse pattern. Lesser the HOMO-LUMO gap means small excitation energy [34]. It was found that the HOMO-LUMO gap for the ligand 97, 109, 48 and 101 in singlet state is 0.23223, 0.1402, 0.15806 and 0.31534 (Graph 3), while in triplet state is 0.00895, 0.01272, 0.02807 and -0.01669. It reveals a good agreement with docking result to be molecule to be enough hard in the binding pocket of protein. The chemical potential (µ) explains the ability of an electron to leave from the molecule in the equilibrium state [35]. The calculated chemical potential for ligand 97 in singlet state and triplet state is found to be 0.11611 and 0.00447 respectively. A significant decrease in chemical potential in triplet state is observed. The chemical hardness of ligand 97, 109, 48 and 101 in singlet state is found to be 0.11611, 0.0701, 0.07903 and 0.15767 respectively, which reveals the hardness order as 101>97>48>109. The global softness value for ligand 97 is found to be 4.30609 and 111.731 in singlet and triplet state respectively. The less hardness value and more softness value makes molecule enough soft and polarizable and the order is 48>109>97>101. The absolute electronegativity is the ability to attract electrons towards itself in a covalent bond [35]. The overall electronegativity of ligand 97, 109, 48 and 101 in singlet state is found to be 0.14192, 0.1518, 0.14183and0.16961respectively and itis less than one, indicate lower electron attraction power of the molecule. Global electrophilicity index indicates the behaviour of molecules to accept the electron density [36]. The electrophilicity value in singlet state is found to be 0.08673, 0.16436, 0.127267and 0.091227 respectively for ligand 97, 109, 48 and 101. The values of all chemical descriptors are given in table 7.
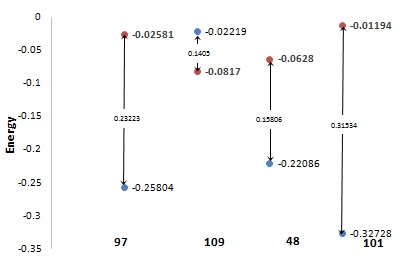
Graph 3: Energies gap of orbitals of HOMO and LUMO of top four
|
|
C. N. 97 |
C. N. 109 |
C. N. 48 |
C. N. 101 |
||||
|
SINGLET |
TRIPLET |
SINGLET |
TRIPLET |
SINGLET |
TRIPLET |
SINGLET |
TRIPLET |
|
|
LUMO+2 |
-0.01399 |
-0.01387 |
-0.06332 |
-0.02612 |
-0.02573 |
-0.02197 |
-0.06943 |
-0.10334 |
|
LUMO+1 |
-0.01686 |
-0.01399 |
-0.06898 |
-0.06332 |
-0.03473 |
-0.02573 |
-0.02863 |
-0.06943 |
|
LUMO |
-0.02581 |
-0.01686 |
-0.0817 |
-0.06898 |
-0.0628 |
-0.03473 |
-0.01194 |
-0.02863 |
|
HOMO |
-0.25804 |
-0.02581 |
-0.2219 |
-0.0817 |
-0.22086 |
-0.0628 |
-0.32728 |
-0.01194 |
|
HOMO-1 |
-0.26057 |
-0.25804 |
-0.2265 |
-0.2219 |
-0.22566 |
-0.22086 |
-0.33317 |
-0.32728 |
|
HOMO-2 |
-0.26867 |
-0.26057 |
-0.22818 |
-0.2265 |
-0.22587 |
-0.22566 |
-0.34232 |
-0.33317 |
|
L-H |
0.23223 |
0.00895 |
0.1402 |
0.01272 |
0.15806 |
0.02807 |
0.31534 |
-0.01669 |
|
L+H |
-0.28385 |
-0.04267 |
-0.3036 |
-0.15068 |
-0.28366 |
-0.09753 |
-0.33922 |
-0.04057 |
|
? |
0.11611 |
0.00447 |
0.0701 |
0.004475 |
0.07903 |
0.014035 |
0.15767 |
0.014035 |
|
Χ |
0.14192 |
0.02133 |
0.1518 |
0.07534 |
0.14183 |
0.048765 |
0.16961 |
0.020285 |
|
S |
4.30607 |
111.731 |
7.132668 |
111.7318 |
6.326711 |
35.62522 |
3.17118 |
35.62522 |
|
µ |
-0.14193 |
-0.02134 |
-0.1518 |
-0.07534 |
-0.14183 |
-0.04877 |
-0.16961 |
0.02029 |
|
Ω |
0.08673 |
0.05085 |
0.16436 |
0.634203 |
0.127267 |
0.084718 |
0.091227 |
0.014659 |
Table 7: Energies of various HOMO, LUMO, and chemical descriptors
The frontier molecular orbital analysis was also done to study the electronic distribution of the electron throughout the molecule. The HOMO and LUMO of ligand 97, 109, 48, and 101are given in figure 2. The HOMO orbital for the ligand 97,iscentered at the nitrogen of isoquinoline ring and the LUMO orbital is at benzofuran ring of noscapine; for ligand 109, the HOMO located on isoquinoline ring and LUMO on substituent ring; for ligand 48, the HOMO located on isoquinoline ring and LUMO on benzofuran ring and for ligand 101, HOMO located on isoquinoline ring and LUMO on substituent ring.

Fig 2: a-e and g represent the frontier molecular orbital of HOMO and b-f and h represent the frontier molecular orbital of LUMO of compound 97, 109, 48, and 101 respectively.
Molecular dynamics simulations analysis
Molecular dynamic simulation is considered to be an important tool to study the behavior of protein as well as protein-ligand complex in the context of structural stability. Herein, the strength, salvation, and conformational pattern are studied. MD simulation of the arginase II with and without the screened ligand was performed using the appropriate force field along with including the explicit solvent [37]. Radius of gyration is an indicator of protein structure’s compactness over the timescale [38]. Rg graph showed slight unfolding in initial time frame but the difference in Rg value of arginase-II with and without 97 is less than the 1.5. But, after a run of 5 ns, it again showed the stable folding and become stable as in graph 4 (a). RMSD measures the similarity in structure of the protein with and without ligand [39]. RMSD of arginase II is approximately found to be the 0.15 nm and for arginase II-97 complex is 0.20 nm graph 4 (b). The RMSD of arginase II and arginase II-97 complex is less than the 0.2 nm and confirms the successful docking.
The root mean square fluctuation (RMSF) is able to compare the fluctuation in the mean position of the backbone atoms of protein [40]. The RMSF values for arginase-II with and without 97 is ranges near the 0.1 nm. It showed the small fluctuations of the atomic coordinates for the protein-ligand complex with reference to the protein ?-carbons. These fluctuations range from 0-6000 atoms of the proteins graph 4 (c). These initial and small fluctuations also support the successful docking of ligand in the active cavity of protein as the cavity amino acid residues belong to 100-300 sequences. Number of H-bonds present between 97 and arginase II is found to 5 in number. Among five three are conventional H-bonds while two are non-conventional H-bonds as in as in graph 4 (d) [41-45].
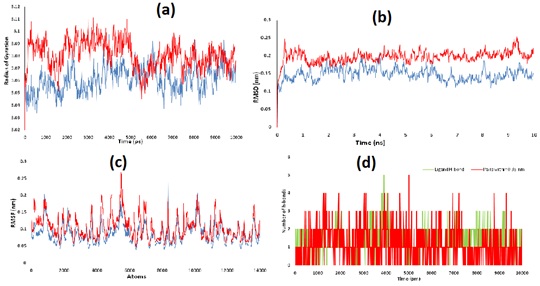 Graph4: (a) Rg behaviour of Arginase-II and Arginase II-97 complex, (b) RMSD behaviour of Arginase-II and Arginase II-97 complex and (c) RMSF behaviour of Arginase-II and Arginase II-97 complex (d) H-bonding interaction of ligand along with pairs within 0.35 nm
Graph4: (a) Rg behaviour of Arginase-II and Arginase II-97 complex, (b) RMSD behaviour of Arginase-II and Arginase II-97 complex and (c) RMSF behaviour of Arginase-II and Arginase II-97 complex (d) H-bonding interaction of ligand along with pairs within 0.35 nm
MM-PBSA analysis
The MD trajectories with no PBC obtained from GROMACS was used to analyze the binding energy, solvation energy and electrostatic potential energy changes of the protein-ligand complex by g_mmpbsa. APBS program was used by the g_mmpbsa to solve the Poisson-Boltzmann (PB) equation. The values of binding energy, SASA, SAV, WCA, van der Waal energy, electrostatic energy and polar solvation along with the maximum possible error in energy are given in table 8. The value of binding energy for the ligand 97 was found to be -815.184 KJ/mol, which is much more negative to support the strong binding of the ligand into the active cavity of protein also show successful docking. The polar solvation energy is enough positive, SASA, van der Waal energy, and electrostatic energies are enough negative.
|
S.N. |
Type of energy |
Value (kJ/mol) |
Error (+/-) |
|
1 |
van der Waal energy |
-131.382 |
13.831 kJ/mol |
|
2 |
Electrostatic energy |
-1135.49 |
50.531 kJ/mol |
|
3 |
Polar solvation energy |
468.828 |
66.349 kJ/mol |
|
4 |
SASA energy |
-17.139 |
1.489 kJ/mol |
|
5 |
SAV energy |
0 |
0.000 kJ/mol |
|
6 |
WCA energy |
0 |
0.000 kJ/mol |
|
7 |
Binding energy |
-815.184 |
32.836 kJ/mol |
Table 8: Results of MM-PBSA analysis of arginase-II-ligand 97 complex
The graph between binding energy, ΔEmm, ΔGpolar, and ΔGnonpolar versus time were also plotted to check the overall response of the system, given in graph 5. The binding energy remains almost invariant around the reported value in table 6, electrostatic potential energy shows more variation in initial time frame, but after 4 ns it also shows almost similar values, the polar solvation energy also initial variation up to 2 ns but after this the values are remains almost constant and non-polar solvation energy initial and final fluctuation but remains almost invariant in range of 2-7 ns.
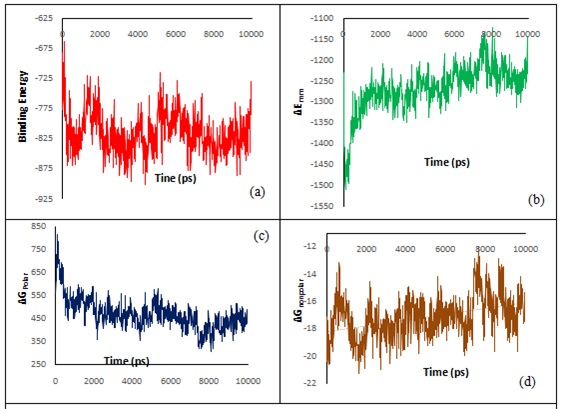
Graph 5: (a) ΔGbinding, (b) ΔEmm, (c) ΔGpolar and (d) ΔGnonpolar versus time of the arginase-II-ligand 97 complex.
CONCLUSION
Herein, the molecular docking, DFT, ADME, MD simulations and MM-PBSA are performed to study the potential of noscapines against the Arginase-II. The potential candidate has been chosen based on binding free energy value due to hydrogen bonding, and van der Waals interaction and electrostatic interaction. Docking analysis shows that ligand 97 have shown the highest binding affinity with arginase-II. Subsequently, the molecular property screening of noscapines satisfied the Lipinski's rule of five. The aqueous solubility of noscapine derivatives was found to be less compared to the reference molecules, and it suggests that it should need to increase. Further, MD simulation is performed to analyze the binding stability of noscapine derivative in the cavity of protein. Rg, RMSD and RMSF results reveal that complex of ligand97 with Arginase-II is highly stable. The high negative value binding energy from mmpbsa results clearly indicates the effective binding of ligand 97witharginase-II. Arginase-II inhibition has been done successfully by ligand 97. This theoretical model was developed to inhibit the arginase II can be used to check the potential of the small molecule against any protein.
REFERENCES
- Benjamin S, Steinhorn, Loscalzo J, Michel T (2015) Nitroglycerin and Nitric Oxide - A Rondo of Themes in Cardiovascular Therapeutics. N Engl J Med 373: 277-280.
- Murrell, W. (1879) Nitroglycerin as a remedy for angina pectoris. Lancet, 1: 80-1.
- Furchgott RF, Zawadzki JV (1980) The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288: 373-376.
- Palmer RM, Ferrige AG & Moncada S (1987) Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327: 524-526.
- Balligand JL, Feron O, Dessy C (2009) eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiological Reviews 80: 481-534.
- Guoyao WU, Sidney MM Jr (1998) Arginine metabolism: Nitric oxide and beyond. Biochem J 336: 1-17.
- Morris SM (2002) Regulation of enzymes of the urea cycle and arginine metabolism. Annu Rev Nutr 22: 87-105.
- Caldwell RB, Toque HA, Narayanan SP, Caldwell RW (2015) Arginase: an old enzyme with new tricks. Trends Pharmacol Sci 36: 395-405.
- Morris SM, Gao T, Cooper TK, Kepka-Lenhart D, Awad AS (2011) Arginase-2 mediates diabetic renal injury. Diabetes 60: 3015-3022.
- Olivon VC, Fraga-Silva RA, Segers D, Demougeot C, de Oliveira AM, et al. (2013) Arginase inhibition prevents the low shear stress-induced development of vulnerable atherosclerotic plaques in Apo E-/- mice. Atherosclerosis 227: 236-243.
- Segal R, Hannan JL, Liu X, Kutlu O, Burnett AL, et al. (2012) Chronic Oral Administration of the Arginase Inhibitor 2(S)-amino-6-boronohexanoic Acid (ABH) Improves Erectile Function in Aged Rats. Journal of Andrology 33: 1169-1175.
- Grasemann H, Dhaliwal R, Ivanovska J, Kantores C, McNamara PJ, et al. (2015) Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am J Physiol Lung Cell Mol Physiol 308: 503-510.
- Bagnost T, Ma L, da Silva RF, Rezakhaniha R, Houdayer C, et al. (2010) Cardiovascular effects of arginase inhibition in spontaneously hypertensive rats with fully developed hypertension. Cardiovasc Res 87: 569-577.
- Meurs H, Zaagsma J, Maarsingh H, vanDuin M (2010) Use of Arginase Inhibitors in the Treatment of Asthma and Allergic Rhinitis. 20150164930 A1. US. 2010.
- Chen X, Dang TT, Facchini PJ (2015) Noscapine comes of age. Phytochemistry 111: 7-13.
- Singh H, Singh P, Kumari K, ChandraA, Dass SK, et al. (2013) A Review on Noscapine, and its Impact on Heme Metabolism. Current drug metabolism 14: 351-360.
- Yang JM, Chen CC (2004) GEMDOCK: a generic evolutionary method for molecular docking. Proteins: Structure, Function and Bioinformatics 55: 288-304.
- Dassault Systèmes BIOVIA, Discovery Studio Modeling Environment, Release 2017, San Diego: Dassault Systèmes, 2016.
- Daina A, Michielin O, Zoete V (2017) Swiss ADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Scientific Reports 7: 42717.
- Zhao Y, Abraham MH, Lee J, Hersey A, Luscombe NC, et al. (2002) Rate-limited steps of human oral absorption and QSAR studies. Pharm Res 19: 1446-1457.
- http://www.molinspiration.com/
- Lagunin A, Zakharov A, Filimonov D, Poroikov V (2011) QSAR Modelling of Rat Acute Toxicity on the Basis of PASS Prediction. Mol Inform 30: 241-250.
- Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, et al. (2009) Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT.
- Bourass M, Benjelloun AT, Benzakour M, Mcharfi M, Hamidi M, et al. (2016) DFT and TD-DFT calculation of new thienopyrazine-based small molecules for organic solar cells. Chemistry Central Journal 10: 67.
- Vishvakarma VK, Singh P, Kumari K, Chandra R (2017) Rational Design of Threo as Well ErythroNoscapines, an Anticancer Drug: A Molecular Docking and Molecular Dynamic Approach. Biochem Pharmacol 6: 229.
- Spoel DVD, Lindahl E, Hess B, Groenhof G, Mark AE, et al (2005) GROMACS: fast, flexible, and free. J Comput Chem 26: 1701-18.
- Vanommeslaeghe K, Hatcher E, Acharya C, Kundu S, Zhong S, et al. (2010) CHARMM general force field: A force field for drug?like molecules compatible with the CHARMM all?atom additive biological force fields. J Comput Chem 31: 671-690.
- Zoete V, Cuendet MA, Grosdidier A, Michielin O (2011) Swiss Param: a fast force field generation tool for small organic molecules. Journal of Computational Chemistry 32: 2359-2368.
- Kumari R (2014) g_mmpbsa - A GROMACS tool for high-throughput MM-PBSA calculations. J Chem Inf Model 54: 1951-1962.
- Baker NA, Sept D, Joseph S, Holst MJ, Andrew Mc Cammon J (2001) Electrostatics of nano systems: Application to microtubules and the ribosome. National Academy of Sciences 98: 10037-10041.
- Vishvakarma VK, Patel R, Kumari K, Singh P (2017) Interaction between Bovine Serum Albumin and Gemini Surfactants using Molecular Docking Characterization. Inf Sci Lett 3: 1-9.
- https://www.cambridgemedchemconsulting.com/resources/ADME/
- Lipinski CA (2004) Lead- and drug-like compounds: the rule-of-five revolution.Drug Discov Today Technol 1: 337–341.
- Pearson RG (1986) Absolute electronegativity and hardness correlated with molecular orbital theory. Proc Natl Acad Sci USA 83: 8440-8441.
- Miura K, Kimata F, Watanabe R, Fukuhara C (2018) DFT Study for Supported Pt Catalysts Focusing on the Chemical Potential. e-Journal of Surface Science and Nanotechnology 16: 209-213.
- Chattaraj PK., Giri S (2009) Electrophilicity index within a conceptual DFT framework. Annu Rep Prog. Chem.Sect C: Phys Chem 105: 13-39.
- Berhanu WM, Masunov AE (2014) Chapter 6 - The Atomic Level Interaction of Polyphenols with the Aβ Oligomer Aggregate, A Molecular Dynamic Guidance for Rational Drug Design. Polyphenols in Human Health and Disease 1: 59-70.
- Lobanov MY, Bogatyreva NS, Galzitskaya OV (2008) Radius of gyration as an indicator of protein structure compactness. Mol Biol 42: 623-628.
- Maiorov VN, Crippen GM (1994) Significance of root-mean-square deviation in comparing three-dimensional structures of globular proteins. J Mol Biol 235: 625-634.
- Fuglebakk E, Echave J, Reuter N (2012) Measuring and comparing structural fluctuation patterns in large protein datasets. Bioinformatics 28: 2431-2440.
- Prashant S, Durgesh K, Vijay KV, Parul Y, Abhilash J, et al. (2019) Computational approach to study the synthesis of noscapine and potential of stereoisomers against nsP3 protease of CHIKV. Heliyon 5: e02795.
- Vijay KV, Prashant S, Vinod K, Kamlesh K, Rajan P, et al. (2019) Pyrrolothiazolones as Potential Inhibitors for the nsP2B?nsP3 Protease of Dengue Virus and Their Mechanism of Synthesis. ChemistrySelect 4: 9410-9419.
- Durgesh K, Kamlesh K, Abhilash J, Prashant S (2019) Development of a theoretical model for the inhibition of nsP3 protease of Chikungunya virus using pyranooxazoles. Journal of Biomolecular and Structural Dynamics 1-17.
- Prashant S, Vijay KV, Nidhi S, Reetu, Kamlesh K, et al. (2019) A model to study the inhibition of nsP2B-nsP3 protease of dengue virus with imidazole, oxazole, triazole thiadiazole, and thiazolidine based scaffolds. Heliyon 5: e02124.
- Durgesh K, Prashant S, Abhilash J, Vinod K, Kamlesh K, et al. (2019) A Theoretical Model to Study the Interaction of Erythro?Noscapines with nsP3 protease of Chikungunya Virus. ChemistrySelect 4: 4892-4900.
SUPPLIMENTARY TABLES
|
Ligand |
Total Energy |
EVDW |
EH-bonding |
EElect |
|
1 |
-91.0327 |
-77.5735 |
-13.4592 |
0 |
|
2 |
-109.384 |
-91.9854 |
-17.3988 |
0 |
|
3 |
-97.4526 |
-85.0978 |
-12.3548 |
0 |
|
4 |
-117.05 |
-94.7327 |
-22.3173 |
0 |
|
5 |
-122.715 |
-89.8135 |
-30.8757 |
-2.02593 |
|
6 |
-96.8304 |
-67.0809 |
-29.7495 |
0 |
|
7 |
-98.741 |
-81.2675 |
-17.4735 |
0 |
|
8 |
-100.539 |
-82.5791 |
-17.9601 |
0 |
|
9 |
-108.578 |
-93.9345 |
-14.6432 |
0 |
|
10 |
-107.478 |
-89.345 |
-18.1333 |
0 |
|
11 |
-106.607 |
-91.8395 |
-14.3133 |
-0.45392 |
|
12 |
-91.975 |
-75.9856 |
-15.9894 |
0 |
|
13 |
-112.295 |
-83.9152 |
-28.3796 |
0 |
|
14 |
-91.8925 |
-91.6397 |
-0.2528 |
0 |
|
15 |
-95.0031 |
-70.1665 |
-24.8366 |
0 |
|
16 |
-104.709 |
-79.8509 |
-24.8586 |
0 |
|
17 |
-97.5492 |
-87.2843 |
-10.2649 |
0 |
|
18 |
-97.2666 |
-75.1413 |
-22.1253 |
0 |
|
19 |
-110.528 |
-94.5839 |
-15.9438 |
0 |
|
20 |
-103.258 |
-85.9429 |
-17.3153 |
0 |
|
21 |
-109.314 |
-93.1677 |
-16.1459 |
0 |
|
22 |
-113.565 |
-87.7818 |
-25.7827 |
0 |
|
23 |
-112.745 |
-94.6705 |
-18.0743 |
0 |
|
24 |
-107.463 |
-104.963 |
-2.5 |
0 |
|
25 |
-112.129 |
-72.67 |
-39.3759 |
-0.0833 |
|
26 |
-107.937 |
-83.6258 |
-24.3114 |
0 |
|
27 |
-120.229 |
-98.5119 |
-21.7173 |
0 |
|
28 |
-102.892 |
-91.5625 |
-11.3293 |
0 |
|
29 |
-115.049 |
-107.605 |
-7.44435 |
0 |
|
30 |
-110.748 |
-98.2447 |
-12.5038 |
0 |
|
31 |
-119.084 |
-98.0764 |
-22.1457 |
1.13783 |
|
32 |
-114.935 |
-98.4504 |
-16.4849 |
0 |
|
33 |
-112.175 |
-99.6831 |
-12.4918 |
0 |
|
34 |
-105.387 |
-85.7027 |
-19.6847 |
0 |
|
35 |
-106.499 |
-88.5389 |
-17.9598 |
0 |
|
36 |
-110.126 |
-95.444 |
-14.6819 |
0 |
|
37 |
-113.429 |
-103.794 |
-9.63506 |
0 |
|
38 |
-117.84 |
-103.04 |
-14.7997 |
0 |
|
39 |
-107.051 |
-96.5513 |
-10.5 |
0 |
|
40 |
-111.64 |
-96.197 |
-15.4426 |
0 |
|
41 |
-112.313 |
-92.4805 |
-19.8324 |
0 |
|
42 |
-107.339 |
-99.6366 |
-7.70222 |
0 |
|
43 |
-104.554 |
-86.3956 |
-18.1585 |
0 |
|
44 |
-121.294 |
-111.479 |
-9.81486 |
0 |
|
45 |
-121.13 |
-99.5557 |
-21.5738 |
0 |
|
46 |
-95.8639 |
-89.139 |
-6.72494 |
0 |
|
47 |
-105.94 |
-95.7799 |
-10.1601 |
0 |
|
48 |
-126.677 |
-98.9796 |
-27.6971 |
0 |
|
49 |
-116.817 |
-107.396 |
-9.42086 |
0 |
|
50 |
-121.996 |
-91.9938 |
-32.3341 |
2.33227 |
|
51 |
-122.622 |
-100.795 |
-21.827 |
0 |
|
52 |
-114.65 |
-105.714 |
-8.93619 |
0 |
|
53 |
-112.345 |
-101.326 |
-11.0192 |
0 |
|
54 |
-108.775 |
-91.8634 |
-16.9117 |
0 |
|
55 |
-110.929 |
-92.4983 |
-18.4306 |
0 |
|
56 |
-112.913 |
-102.64 |
-10.2734 |
0 |
|
57 |
-116.385 |
-103.495 |
-12.8901 |
0 |
|
58 |
-115.883 |
-105.239 |
-10.644 |
0 |
|
59 |
-108.636 |
-98.431 |
-10.2054 |
0 |
|
60 |
-106.02 |
-92.5062 |
-13.5141 |
0 |
|
61 |
-105.844 |
-99.9673 |
-5.87681 |
0 |
|
62 |
-109.801 |
-98.291 |
-11.5098 |
0 |
|
63 |
-112.478 |
-88.3698 |
-22.5875 |
-1.52103 |
|
64 |
-116.888 |
-89.5448 |
-27.3432 |
0 |
|
65 |
-116.913 |
-90.9412 |
-25.9715 |
0 |
|
66 |
-104.905 |
-93.8031 |
-11.1022 |
0 |
|
67 |
-108.323 |
-78.5167 |
-29.8065 |
0 |
|
68 |
-122.707 |
-104.85 |
-17.8573 |
0 |
|
69 |
-120.698 |
-107.083 |
-13.1938 |
-0.42128 |
|
70 |
-104.602 |
-88.7713 |
-15.8304 |
0 |
|
71 |
-118.534 |
-102.436 |
-16.0977 |
0 |
|
72 |
-109.145 |
-90.5591 |
-18.5855 |
0 |
|
73 |
-100.979 |
-88.2069 |
-12.7722 |
0 |
|
74 |
-105.235 |
-84.2613 |
-20.9739 |
0 |
|
75 |
-104.475 |
-95.7479 |
-8.72682 |
0 |
|
76 |
-101.485 |
-87.2427 |
-14.242 |
0 |
|
77 |
-110.902 |
-94.2911 |
-16.6108 |
0 |
|
78 |
-113.671 |
-106.78 |
-6.89113 |
0 |
|
79 |
-118.162 |
-91.5669 |
-26.5951 |
0 |
|
80 |
-111.439 |
-98.9876 |
-12.4519 |
0 |
|
81 |
-102.866 |
-94.1186 |
-8.74766 |
0 |
|
82 |
-119.63 |
-74.5102 |
-43.7129 |
-1.40655 |
|
83 |
-115 |
-87.7448 |
-27.255 |
0 |
|
84 |
-111.556 |
-85.2251 |
-26.3308 |
0 |
|
85 |
-111.459 |
-92.2666 |
-19.1923 |
0 |
|
86 |
-113.328 |
-100.878 |
-12.4498 |
0 |
|
87 |
-108.786 |
-80.5001 |
-28.2856 |
0 |
|
88 |
-121.07 |
-80.7958 |
-40.764 |
0.489526 |
|
89 |
-112.954 |
-102.484 |
-10.4704 |
0 |
|
90 |
-113.741 |
-101.624 |
-12.1168 |
0 |
|
91 |
-108.255 |
-87.3362 |
-20.9189 |
0 |
|
92 |
-105.69 |
-97.7104 |
-7.97946 |
0 |
|
93 |
-115.311 |
-107.016 |
-8.29515 |
0 |
|
94 |
-116.222 |
-92.1833 |
-24.0391 |
0 |
|
95 |
-124.896 |
-107.441 |
-17.4548 |
0 |
|
96 |
-110.211 |
-103.04 |
-7.1707 |
0 |
|
97 |
-133.413 |
-93.6378 |
-39.7757 |
0 |
|
98 |
-119.453 |
-102.307 |
-17.1458 |
0 |
|
99 |
-109.284 |
-102.616 |
-6.66826 |
0 |
|
100 |
-122.101 |
-107.862 |
-14.2386 |
0 |
|
101 |
-126.298 |
-95.8055 |
-28.7223 |
-1.77013 |
|
102 |
-105.874 |
-89.2876 |
-16.5864 |
0 |
|
103 |
-109.055 |
-94.4301 |
-14.6251 |
0 |
|
104 |
-114.627 |
-99.8451 |
-14.7815 |
0 |
|
105 |
-114.19 |
-101.451 |
-12.7391 |
0 |
|
106 |
-113.111 |
-104.256 |
-8.85546 |
0 |
|
107 |
-115.01 |
-91.0375 |
-22.433 |
-1.53905 |
|
108 |
-113.338 |
-90.8417 |
-22.4964 |
0 |
|
109 |
-129.175 |
-115.069 |
-14.1058 |
0 |
|
110 |
-118.448 |
-104.196 |
-14.2518 |
0 |
|
111 |
-105.64 |
-93.8759 |
-11.7642 |
0 |
|
112 |
-105.872 |
-92.0203 |
-13.8518 |
0 |
|
113 |
-117.354 |
-93.8579 |
-23.4964 |
0 |
|
114 |
-115.442 |
-97.6821 |
-17.7595 |
0 |
|
115 |
-114.528 |
-104.288 |
-10.2402 |
0 |
|
116 |
-119.553 |
-101.666 |
-17.887 |
0 |
Table SA: Docking score of the noscapines
|
Ligand |
Log S |
Solubility |
Log Kp |
Log P |
|
1 |
-4.14 |
0.0298 |
-6.9 |
3.59 |
|
2 |
-3.68 |
0.0919 |
-7.71 |
4 |
|
3 |
-4.99 |
0.00522 |
-7.07 |
4 |
|
4 |
-4.6 |
0.0115 |
-6.92 |
3.86 |
|
5 |
-4.22 |
0.0755 |
-7.29 |
3.01 |
|
6 |
-3.8 |
0.0683 |
-7.47 |
3.31 |
|
7 |
-4.74 |
0.00809 |
-6.66 |
3.71 |
|
8 |
-5.06 |
0.00432 |
-6.89 |
3.82 |
|
9 |
-3.81 |
0.0724 |
-7.83 |
3.6 |
|
10 |
-4.76 |
0.00831 |
-6.92 |
3.65 |
|
11 |
-1.88 |
6 |
-9.12 |
3.08 |
|
12 |
-3.89 |
0.0056 |
-7.45 |
3.08 |
|
13 |
-3.98 |
0.0487 |
-7.38 |
3.61 |
|
14 |
-4.7 |
0.00886 |
-6.51 |
3.88 |
|
15 |
-4.45 |
0.0153 |
-6.73 |
3.81 |
|
16 |
-4.23 |
0.0264 |
-7.01 |
3.92 |
|
17 |
-4.47 |
0.0155 |
-6.93 |
3.87 |
|
18 |
-4.01 |
0.0424 |
-7.25 |
3.37 |
|
19 |
-4.11 |
0.0355 |
-7.38 |
3.61 |
|
20 |
-4.1 |
0.0346 |
-7.25 |
3.45 |
|
21 |
-5.65 |
0.0011 |
-6.2 |
4.11 |
|
22 |
-5.19 |
0.00336 |
-7.02 |
4.3 |
|
23 |
-6.49 |
0.00019 |
-6.38 |
4.37 |
|
24 |
-6.1 |
0.000424 |
-6.23 |
4.28 |
|
25 |
-5.72 |
0.00102 |
-6.61 |
3.86 |
|
26 |
-5.3 |
0.00254 |
-6.79 |
3.85 |
|
27 |
-6.24 |
0.000299 |
-5.97 |
4.4 |
|
28 |
-6.56 |
0.000155 |
-6.2 |
4.5 |
|
29 |
-5.31 |
0.00267 |
-7.14 |
4.04 |
|
30 |
-6.26 |
0.000305 |
-6.23 |
4.11 |
|
31 |
-4.9 |
0.0431 |
-8.43 |
3.24 |
|
32 |
-5.4 |
0.00207 |
-6.76 |
3.84 |
|
33 |
-5.61 |
0.00129 |
-6.69 |
4.09 |
|
34 |
-6.21 |
0.000318 |
-5.82 |
4.4 |
|
35 |
-5.95 |
0.000561 |
-6.03 |
4.33 |
|
36 |
-5.73 |
0.000965 |
-6.41 |
4.39 |
|
37 |
-5.97 |
0.000572 |
-6.24 |
4.4 |
|
38 |
-5.51 |
0.00156 |
-6.56 |
3.9 |
|
39 |
-6.58 |
0.000158 |
-6.45 |
4.21 |
|
40 |
-5.6 |
0.00129 |
-6.56 |
4.07 |
|
41 |
-5.19 |
0.00336 |
-7.02 |
4 |
|
42 |
-6.49 |
0.00019 |
-6.38 |
4.4 |
|
43 |
-6.1 |
0.000424 |
-6.23 |
4.38 |
|
44 |
-5.72 |
0.00102 |
-6.61 |
3.88 |
|
45 |
-5.3 |
0.00254 |
-6.79 |
3.79 |
|
46 |
-6.24 |
0.000299 |
-5.97 |
4.5 |
|
47 |
-6.56 |
0.000155 |
-6.2 |
4.45 |
|
48 |
-5.31 |
0.00267 |
-7.14 |
4 |
|
49 |
-6.26 |
0.000305 |
-6.23 |
3.98 |
|
50 |
-4.09 |
0.0435 |
-8.43 |
3.86 |
|
51 |
-5.4 |
0.00207 |
-6.76 |
4 |
|
52 |
-5.61 |
0.00129 |
-6.69 |
3.95 |
|
53 |
-6.21 |
0.000318 |
-5.82 |
4.4 |
|
54 |
-5.59 |
0.000561 |
-6.03 |
4.5 |
|
55 |
-5.73 |
0.000965 |
-6.41 |
4.08 |
|
56 |
-5.97 |
0.000572 |
-6.24 |
4.77 |
|
57 |
-5.51 |
0.00156 |
-6.56 |
3.92 |
|
58 |
-6.58 |
0.000158 |
-6.45 |
3.96 |
|
59 |
-5.6 |
0.00129 |
-6.56 |
4.19 |
|
60 |
-5.19 |
0.00336 |
-7.02 |
4.19 |
|
61 |
-6.49 |
0.00019 |
-6.38 |
4.51 |
|
62 |
-6.1 |
0.000424 |
-6.23 |
4.35 |
|
63 |
-5.72 |
0.00102 |
-6.61 |
3.72 |
|
64 |
-5.3 |
0.00254 |
-6.79 |
3.82 |
|
65 |
-6.24 |
0.000299 |
-5.97 |
4.5 |
|
66 |
-5.56 |
0.000155 |
-6.2 |
4.58 |
|
67 |
-5.31 |
0.00267 |
-7.14 |
3.99 |
|
68 |
-6.26 |
0.000305 |
-6.23 |
4.22 |
|
69 |
-4.09 |
0.0435 |
-8.43 |
3.67 |
|
70 |
-5.4 |
0.00207 |
-6.76 |
3.87 |
|
71 |
-5.91 |
0.00129 |
-6.69 |
4.09 |
|
72 |
-6.21 |
0.000318 |
-5.82 |
4.37 |
|
73 |
-5.95 |
0.000561 |
-6.03 |
4.42 |
|
74 |
-5.73 |
0.000965 |
-6.41 |
4.37 |
|
75 |
-5.97 |
0.000572 |
-6.24 |
4.68 |
|
76 |
-5.51 |
0.00156 |
-6.56 |
3.86 |
|
77 |
-6.58 |
0.000158 |
-6.45 |
4.32 |
|
78 |
-5.6 |
0.00129 |
-6.56 |
4.15 |
|
79 |
-4.73 |
0.0102 |
-7.83 |
4.11 |
|
80 |
-7.33 |
0.0000315 |
-6.55 |
4.54 |
|
81 |
-6.57 |
0.000159 |
-6.25 |
4.39 |
|
82 |
-5.8 |
0.000915 |
-7 |
3.5 |
|
83 |
-4.95 |
0.00579 |
-7.36 |
3.63 |
|
84 |
-6.84 |
0.0000799 |
-5.74 |
4.49 |
|
85 |
-7.48 |
0.00001216 |
-6.19 |
4.55 |
|
86 |
-4.99 |
0.00624 |
-8.07 |
3.93 |
|
87 |
-6.88 |
0.0000819 |
-6.25 |
4.03 |
|
88 |
-3.98 |
0.0612 |
-9.03 |
2.95 |
|
89 |
-5.15 |
0.00389 |
-7.31 |
3.64 |
|
90 |
-5.58 |
0.0015 |
-7.17 |
3.81 |
|
91 |
-6.78 |
0.00009 |
-5.42 |
4.58 |
|
92 |
-6.26 |
0.000286 |
-5.86 |
4.37 |
|
93 |
-5.81 |
0.000842 |
-6.61 |
4.32 |
|
94 |
-6.3 |
0.000289 |
-6.27 |
4.91 |
|
95 |
-5.38 |
0.00217 |
-6.9 |
3.87 |
|
96 |
-7.51 |
0.0000218 |
-6.7 |
3.99 |
|
97 |
-5.56 |
0.00148 |
-6.91 |
3.9 |
|
98 |
-4.73 |
0.0102 |
-7.83 |
4.29 |
|
99 |
-7.33 |
0.0000315 |
-6.5 |
5.02 |
|
100 |
-6.57 |
0.000159 |
-6.25 |
4.41 |
|
101 |
-5.8 |
0.000915 |
-7 |
3.18 |
|
102 |
-4.95 |
0.00579 |
-7.36 |
3.45 |
|
103 |
-6.84 |
0.0000799 |
-7.36 |
3.45 |
|
104 |
-7.48 |
0.0000216 |
-6.19 |
4.78 |
|
105 |
-4.99 |
0.00624 |
-8.07 |
3.75 |
|
106 |
-6.88 |
0.0000819 |
-6.25 |
4.08 |
|
107 |
-3.98 |
0.0612 |
-9.03 |
3.12 |
|
108 |
-5.15 |
0.00389 |
-7.31 |
3.71 |
|
109 |
-5.58 |
0.0015 |
-7.17 |
4 |
|
110 |
-6.78 |
0.00009 |
-5.42 |
4.81 |
|
111 |
-6.26 |
0.000286 |
-5.86 |
4.64 |
|
112 |
-5.81 |
0.000842 |
-6.61 |
4.51 |
|
113 |
-6.3 |
0.0002.89 |
-6.27 |
5 |
|
114 |
-7.51 |
0.0000218 |
-6.7 |
4.03 |
|
115 |
-5.56 |
0.00148 |
-6.91 |
3.9 |
Table SB: ADME properties of the designed noscapines
Citation: Vishvakarma VK, Singh P, Kumari K (2020) A model to study the inhibition of Arginase II with Noscapine & its derivatives. J Protein Res Bioinform 2: 008.
Copyright: © 2020 Vijay Kumar Vishvakarma2,, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.