A New Role of the Proteasome in Alpha One Antitrypsin Regulation
*Corresponding Author(s):
Lang RaoSouthern California Institute For Research And Education, VA Long Beach Healthcare System, Long Beach, CA 90822, United States
Tel:+1 5628268000 (ext.15676),
Email:lang.rao@va.gov
Abstract
In this study, we showed that proteasome inhibitors selectively suppress the synthesis of Alpha One Antitrypsin (α1AT) by inducing phosphorylation of eukaryotic translation initiation factor 2α and increasing the association of stress granule RNA binding proteins with α1AT mRNA. Further, we showed that proteasome inhibitors could also cause selective translational inhibition of mutant α1AT in induced pluripotent stem cell-derived hepatocytes from patients with α1AT deficiency. Thus, our study reveals that the proteasome is not only responsible for α1ATdegradation, it also regulates α1AT translation.
Keywords
Alpha one antitrypsin; Alpha one antitrypsin deficiency; Induced pluripotent stem cells; Proteasome
INTRODUCTION
Alpha One Antitrypsin (α1AT), encoded by the gene SERPINA1, is a glycoprotein synthesized by the liver and functions as an elastase inhibitor in the lung, where it protects the pulmonary tissues from elastase-mediated degradation. Mutation in SERPINA1 results in the retention of nonfunctional misfolded α1AT mutant protein inside liver cells leading to the deficiency of α1AT in circulation, thereby exposing the lung to the elastase-mediated digestion [1,2]. This SERPINA1gene mutation resulted symptom is called Alpha One Antitrypsin Deficiency (α1ATD). Patients with α1ATD often develop chronic obstructive pulmonary disorder and liver cirrhosis. Though Intravenous α1AT augmentation therapy is available to alleviate lung damage, there is very limited effective therapy to mitigate liver damage which leaves the patient with liver transplantation as the only option [3]. Different strategies have been tried to reduce α1ATD liver damage. These methods focused on suppressing α1AT mRNA transcription, removing aggregated proteins and blocking the mutant protein polymerization [4]. Even though suppressing mutant α1AT synthesis could be a practical strategy to alleviate liver toxicity, there has been little focus on this area of research. Our study started with the finding that PS-341, an FDA approved anticancer drug [5], has a strong inhibitory effect on the synthesis of α1AT in human hepatocytes. As PS-341 usually acts as a typical proteasome inhibitor, it thus suggested that proteasome inhibition could lead to α1AT synthesis suppression. Other proteasome antagonists like MG132, lactacystin, and epoxomicin could all cause significant inhibition of α1AT expression, thereby supporting the notion that proteasome inhibition suppresses the production of α1AT.
By comparing the protein synthesis level using isotope labeling and polyribosome profiling assays we showed that proteasome inhibitor MG132 selectively suppressed α1AT translation. It significantly affected the translation of α1AT whereas the general protein synthesis remained nearly unchanged [6]. To identify how the proteasome inhibitors selectively suppressed α1AT translation, we analyzed different signaling pathways involved in translational control of protein synthesis. We found a correlation of eukaryotic translation initiation factor 2 α (eiF2α) phosphorylation with the decreased expression of α1AT. Besides, overexpression of eiF2α specific phosphatase GADD34 could partially restore the α1AT translational inhibition, indicating that eIF2α phosphorylation contributes to the inhibition of α1AT protein synthesis. However, a typical ER stress inducer thapsigargin caused robust phosphorylation of eIF2α but only a slight inhibition of α1AT, indicating that proteasome inhibitor-mediated α1AT inhibition only partially depends on eIF2α phosphorylation [6].
In different cells, proteasome inhibitors could induce stress granules and mediate mRNA translation inhibition [7]. In our study, we also observed that proteasome inhibitors induced stress granule formation in hepatocytes, and importantly we found an increased association of RNA Binding Proteins (RBPs) with α1AT mRNA. Overexpression of one of the identified RBPs G3BP1 could enhance proteasome inhibitor-induced α1AT translational repression. It thus indicated that proteasome inhibitors induce stress granule formation and association of RBPs with α1AT to suppress mRNA translation. Overall, we propose that proteasome inhibitors inhibit the translation of α1AT, which is a combinatorial effect of eIF2α phosphorylation and stress granule protein association (Figure 1) [6]. This selective inhibition of proteins synthesis by proteasome inhibitors could also apply to other hepatic secretory proteins because we have also observed a similar phenomenon on albumin and fetoprotein [6].
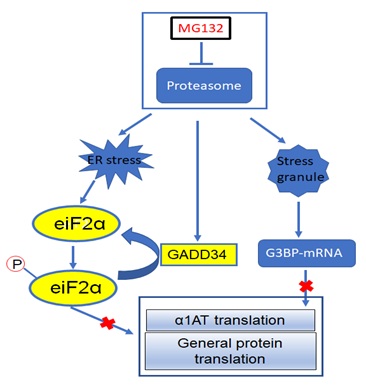 Figure 1: Mechanism of proteasome inhibition regulates α1AT translation.
Figure 1: Mechanism of proteasome inhibition regulates α1AT translation.
After showing the effects of proteasome inhibitors on translational inhibition of wild type α1AT, we tried to identify if the mutant α1AT could also be affected similarly. We used the same proteasome inhibitors on induced Pluripotent Stem Cells (iPSC) derived hepatocytes from α1ATD patients. We found that the translation of mutant α1AT in iPSC derived hepatocytes was also downregulated following proteasome inhibition. Previous research showed that the proteasome is partially responsible for degrading the intracellular mutant α1AT aggregates [8], our funding that proteasome inhibitors suppress mutant α1AT synthesis provides a new hint on drug development for α1ATD treatment.
Proteasome inhibitors inhibits the proteasome and induce ER Stress. During ER stress, ER-resident transmembrane proteins (PERK) activate phosphorylation of the eIF2α and represses general protein translation. Proteasome inhibition also triggers the accumulation of eIF2α specific phosphatase GADD34 which attenuates eIF-2α phosphorylation. Meanwhile, proteasome inhibitors caused the formation of stress granules which recruit the α1AT mRNA and related RNA binding proteins like G3BP1 and prevent the mRNA from translation. As a whole, α1AT translation was severely inhibited by proteasome inhibitor.
REFERENCES
- Stoller JK, Aboussouan LS (2012) A review of alpha1-antitrypsin deficiency. Am J Respir Crit Care Med 185: 246-259.
- Fairbanks KD, Tavill AS (2008) Liver disease in alpha 1-antitrypsin deficiency: a review. Am J Gastroenterol 103: 2136-2141
- Mohanka M, Khemasuwan D, Stoller JK (2012) A review of augmentation therapy for alpha-1 antitrypsin deficiency. Expert Opin Biol Ther 12: 685-700.
- Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, et al. (2010) An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science 329: 229-232.
- Chen D, Frezza M, Schmitt S, Kanwar J, Dou QP (2011) Bortezomib as the first proteasome inhibitor anticancer drug: current status and future perspectives. Curr Cancer Drug Targets 11: 239-253.
- Rao L, Xu Y, Reineke LC, Bhattacharya A, Tyryshkin A, et al. (2020) Post-Transcriptional Regulation of Alpha One Antitrypsin by a Proteasome Inhibitor. Int J Mol Sci 21: 4318.
- Mazroui R, Di Marco S, Kaufman RJ, Gallouzi I-E (2007) Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol Biol Cell 18: 2603-2618.
- Teckman JH, Burrows J, Hidvegi T, Schmidt B, Hale PD, et al. (2001) The proteasome participates in degradation of mutant alpha 1-antitrypsin Z in the endoplasmic reticulum of hepatoma-derived hepatocytes. J Biol Chem 276: 44865-44872.
Citation: Rao L, Eissa NT (2020) A New Role of the Proteasome in Alpha One Antitrypsin Regulation. J Stem Cell Res Dev Ther 6: 048.
Copyright: © 2020 Lang Rao, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.