Journal of Pulmonary Medicine & Respiratory Research Category: Medical
Type: Case Report
Clinical and Haemodynamic Outcomes of the Macitentan Plus Tadalafil Combination for Severe Portopulmonary Hypertension: First Experience in Routine Clinical Practice
*Corresponding Author(s):
Clara Itzíar Soto AbánadesDepartment Of Internal Medicine, GRUHPAZ (GRupo Multidisciplinar De Hipertensión Pulmonar Del Hospital La Paz), Hospital Universitario La Paz, Madrid, Spain
Tel:+34 917277188 / +34 607453630,
Email:clarasoto27@gmail.com
Received Date: Jun 07, 2018
Accepted Date: Jul 17, 2018
Published Date: Jul 31, 2018
Abstract
A 57-year-old male with a history of chronic liver disease and portal hypertension complicated with oesophageal varices, presented severe portopulmonary hypertension (WHO functional class III). The diagnosis was confirmed after right heart catheterisation (mean Pulmonary Arterial Pressure [mPAP]: 55 mmHg; Pulmonary Vascular Resistance [PVR]: 12.8 WU). After starting ambrisentan plus tadalafil, the patient developed painful oedema in the lower limbs, and ambrisentan was switched to macitentan 10 mg/day. After 3 months of combined treatment, the patient’s functional class improved to WHO II, with no dyspnoea on ordinary exertion and complete resolution of oedema. At 6 months, the patient showed significant decreases in both mPAP (38 mmHg) and PVR (5.7 WU). One year after the start of the combination, the patient presents WHO functional class I, with no dyspnoea and no limitations in his daily activities. In this clinical case, the combination of macitentan plus tadalafil provided rapid and significant improvement of severe portopulmonary hypertension.
Keywords
Macitentan; Portopulmonary hypertension; Tadalafil
INTRODUCTION
Portopulmonary Hypertension (POPH) is a complication observed in 2-6% of patients with portal hypertension, namely in those developing Pulmonary Arterial Hypertension (PAH) [1,2]. POPH is classified as a subtype of Group 1 PAH [3], which is characterized by pulmonary vasoconstriction and pulmonary vascular remodeling associated with endothelial, fibroblast and smooth muscle cell proliferation. It is a severe condition that can lead to right heart failure and death. Differentiation of POPH from other causes of pulmonary hypertension is critical given the significant implications of its diagnosis for liver transplant risk stratification, Model for End-Stage Liver Disease (MELD) exception points, and selection of PAH-specific therapy.
Mainly, the therapies used to treat POPH are vasodilators used in PAH, including monotherapy or combination therapy with prostanoids, endothelin receptor antagonists, and phosphodiesterase-5 inhibitors/guanylate cyclase stimulator [4]. Several new treatments with proven efficacy and safety in PAH have been steadily approved in the past years. However, to date, the evidence of use of these therapies to treat POPH is scarce, and limited to small case series or reports. Here, we report a clinical case of a patient successfully treated with macitentan, a dual endothelin receptor antagonist, plus tadalafil upon diagnosis of severe POPH.
Mainly, the therapies used to treat POPH are vasodilators used in PAH, including monotherapy or combination therapy with prostanoids, endothelin receptor antagonists, and phosphodiesterase-5 inhibitors/guanylate cyclase stimulator [4]. Several new treatments with proven efficacy and safety in PAH have been steadily approved in the past years. However, to date, the evidence of use of these therapies to treat POPH is scarce, and limited to small case series or reports. Here, we report a clinical case of a patient successfully treated with macitentan, a dual endothelin receptor antagonist, plus tadalafil upon diagnosis of severe POPH.
CASE REPORT
A 57-year-old male presented with progressive dyspnoea. He was an active smoker with a previous history of chronic alcoholism up until eight years prior. This had led to chronic liver disease with portal hypertension complicated with oesophageal varices (Child-Pugh class B, 7 points, MELD score 16 points at the time of the visit). He was being monitored by the gastroenterology department and had undergone a TIPS (Transjugular Intrahepatic Portosystemic Shunt) procedure three years earlier as a therapeutic measure. He was not hypertensive nor presented any other typical vascular risk factor, except for smoking. He had no known previous history of heart, cerebrovascular, kidney or peripheral artery disease. He did not meet the clinical criteria for chronic bronchitis. He had no history of surgery, except for the TIPS, and his functional status was excellent: He worked as a manager of a company and had an active life with no physical limitations. His usual treatment included ursodeoxycholic acid, bisoprolol and lactitol.
He was referred from the hepatology unit due to a two-month history dyspnoea that had progressed to dyspnoea on minimal exertion (WHO functional class III). It was not accompanied by cough, expectoration, fever or other infectious symptoms. He reported no palpitations, chest pain or syncope. There was no evidence of constitutional syndrome or haemoptysis. Physical examination showed blood pressure of 120/60 mmHg, a resting heart rate of 70 beats per minute, eupnoea and a baseline oxygen saturation of 95%. Cardiopulmonary auscultation was normal. The abdominal examination revealed mild collateral circulation without ascites, and a 2 cm non-tender hepatomegaly on palpation. Both extremities presented discrete ankle oedema with pitting. Lab tests showed 89,000×103/μl platelets (other complete blood count parameters were normal), aspartate transaminase: 54 IU/l, alanine transaminase: 15 IU/l, gamma-glutamyltransferase: 483 IU/l, bilirubin: 6.4 mg/dl and alkaline phosphatase: 229 IU/l, decreased prothrombin activity (57%), and NT-proBNP (1,841 pg/ml). The abdominal ultrasound showed no changes compared to previous scans, evidencing chronic liver disease with splenomegaly, without ascites, permeable TIPS, portal vein with an enlarged 21.6 mm lumen and a flow rate of 30.5 cm/s. Respiratory function tests ruled out obstructive and restrictive lung disease and identified a reduced (63.4%) diffusing capacity for carbon monoxide (DLCO). The pulmonary ventilation/perfusion scan showed no defects suggestive of chronic pulmonary thromboembolism, and the Computed Tomography (CT) angiography of the pulmonary arteries showed no intravascular filling defects or parenchymal lesion. The patient reached 360 m on the 6-Minute Walk Test (6 MWT), with a decrease in oxygen saturation to 86%. Echocardiography findings were: Severely dilated and hypokinetic Right Ventricle (RV), with Tricuspid Annular Plane Systolic Excursion (TAPSE) of 9 mm and fractional shortening of 16%; severe tricuspid insufficiency with regurgitation velocity of 341 cm/sec, estimating a pulmonary systolic pressure of 66 mmHg; right atrial enlargement, with a four-chamber view of 25.4 cm2. There was a global hypokinesis in the systolic function. The left atrium, the contractility and left ventricular ejection fraction, as well as the other heart valves, were normal (Figure 1A and 1B) (Supplementary Files 1 and 2). The presumptive diagnosis of severe portopulmonary hypertension (WHO functional class III) was confirmed after right heart catheterisation (Table 1) and discarding other causes of pulmonary hypertension.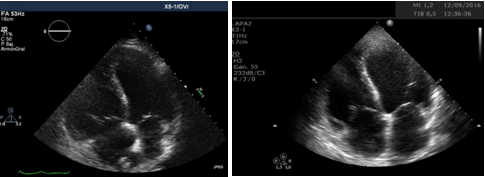 Figure 1: Echocardiography A) At portopulmonary hypertension diagnosis; B) After treatment.Table 1: Right heart catheterisation results before and after treatment.
Figure 1: Echocardiography A) At portopulmonary hypertension diagnosis; B) After treatment.Table 1: Right heart catheterisation results before and after treatment.
He was referred from the hepatology unit due to a two-month history dyspnoea that had progressed to dyspnoea on minimal exertion (WHO functional class III). It was not accompanied by cough, expectoration, fever or other infectious symptoms. He reported no palpitations, chest pain or syncope. There was no evidence of constitutional syndrome or haemoptysis. Physical examination showed blood pressure of 120/60 mmHg, a resting heart rate of 70 beats per minute, eupnoea and a baseline oxygen saturation of 95%. Cardiopulmonary auscultation was normal. The abdominal examination revealed mild collateral circulation without ascites, and a 2 cm non-tender hepatomegaly on palpation. Both extremities presented discrete ankle oedema with pitting. Lab tests showed 89,000×103/μl platelets (other complete blood count parameters were normal), aspartate transaminase: 54 IU/l, alanine transaminase: 15 IU/l, gamma-glutamyltransferase: 483 IU/l, bilirubin: 6.4 mg/dl and alkaline phosphatase: 229 IU/l, decreased prothrombin activity (57%), and NT-proBNP (1,841 pg/ml). The abdominal ultrasound showed no changes compared to previous scans, evidencing chronic liver disease with splenomegaly, without ascites, permeable TIPS, portal vein with an enlarged 21.6 mm lumen and a flow rate of 30.5 cm/s. Respiratory function tests ruled out obstructive and restrictive lung disease and identified a reduced (63.4%) diffusing capacity for carbon monoxide (DLCO). The pulmonary ventilation/perfusion scan showed no defects suggestive of chronic pulmonary thromboembolism, and the Computed Tomography (CT) angiography of the pulmonary arteries showed no intravascular filling defects or parenchymal lesion. The patient reached 360 m on the 6-Minute Walk Test (6 MWT), with a decrease in oxygen saturation to 86%. Echocardiography findings were: Severely dilated and hypokinetic Right Ventricle (RV), with Tricuspid Annular Plane Systolic Excursion (TAPSE) of 9 mm and fractional shortening of 16%; severe tricuspid insufficiency with regurgitation velocity of 341 cm/sec, estimating a pulmonary systolic pressure of 66 mmHg; right atrial enlargement, with a four-chamber view of 25.4 cm2. There was a global hypokinesis in the systolic function. The left atrium, the contractility and left ventricular ejection fraction, as well as the other heart valves, were normal (Figure 1A and 1B) (Supplementary Files 1 and 2). The presumptive diagnosis of severe portopulmonary hypertension (WHO functional class III) was confirmed after right heart catheterisation (Table 1) and discarding other causes of pulmonary hypertension.
Figure 1: Echocardiography A) At portopulmonary hypertension diagnosis; B) After treatment.Table 1: Right heart catheterisation results before and after treatment.
| At Diagnosis | After 6 Months (Macitentan + Tadalafil) | |
| Systolic pulmonary arterial pressure | 93 mmHg | 60 mmHg |
| Diastolic pulmonary arterial pressure | 36 mmHg | 20 mmHg |
| Mean pulmonary arterial pressure | 55 mmHg | 38 mmHg |
| Pulmonary capillary wedge pressure | 14 mmHg | 15 mmHg |
| Right atrial pressure | 20 mmHg | 15 mmHg |
| Pulmonary vascular resistance | 12.8 WU | 5.7 WU |
| Cardiac output | 3.5 l/min | 3.8 l/min |
| Cardiac index | 1.6 l/min/m2 | 2.0 l/min/m2 |
| Transpulmonary gradient | 43 mmHg | 22 mmHg |
| Porto-caval gradient | 5 mmHg | 5 mmHg |
Following diagnosis, the first therapeutic measure was the withdrawal of beta-blockers. We suggested the onset of parenteral prostanoids, but the patient rejected it. Therefore, we decided on combination oral therapy, starting with 5 mg ambrisentan and 20 mg tadalafil daily in order to control possible side effects, and then escalate the dose to 10 mg ambrisentan and 40 mg tadalafil within two weeks [5]. Despite initial improvement of dyspnoea, the patient presented significant, painful oedema in the lower limbs that was refractory to diuretic treatment one month after treatment onset, before reaching the full dose of both drugs. As this was concomitant with the start of treatment, it was decided to discontinue ambrisentan. Tadalafil was increased to the target dose of 40 mg daily, with no adverse effects, and macitentan, a dual endothelin receptor antagonist that presented low hepatotoxicity and low risk of peripheral oedema in the pivotal trial SERAPHIN [6], was started at a dose of 10 mg daily.
Three months after the start of the new combined treatment regimen, the patient’s functional class improved to WHO II, with no dyspnoea on ordinary exertion (ambulation on a flat surface) and complete resolution of peripheral oedema. A complete re-evaluation was performed at six months, with both invasive and non-invasive studies, and a new catheterisation that showed significant decreases in both mean pulmonary pressure and pulmonary vascular resistance, as well as an increase, though less pronounced, in the cardiac index (Table 1). Regarding other prognostic parameters, NT-proBNP decreased to normal levels, 6 MWT increased by 30 metres (Figure 2), and echocardiographic parameters improved (TAPSE: 32 mm, fractional shortening 37%, right atrial area: 22.9 cm2, preserved global systolic function, moderate RV dilatation) (Figures 3A and 3B).
Three months after the start of the new combined treatment regimen, the patient’s functional class improved to WHO II, with no dyspnoea on ordinary exertion (ambulation on a flat surface) and complete resolution of peripheral oedema. A complete re-evaluation was performed at six months, with both invasive and non-invasive studies, and a new catheterisation that showed significant decreases in both mean pulmonary pressure and pulmonary vascular resistance, as well as an increase, though less pronounced, in the cardiac index (Table 1). Regarding other prognostic parameters, NT-proBNP decreased to normal levels, 6 MWT increased by 30 metres (Figure 2), and echocardiographic parameters improved (TAPSE: 32 mm, fractional shortening 37%, right atrial area: 22.9 cm2, preserved global systolic function, moderate RV dilatation) (Figures 3A and 3B).
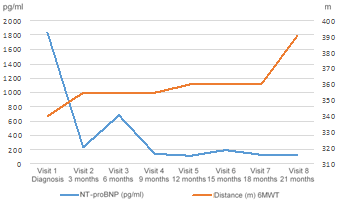 Figure 2: Evolution of NT-proBNP levels and 6-Minute Walk Distance (6 MWD) after portopulmonary hypertension diagnosis.
Figure 2: Evolution of NT-proBNP levels and 6-Minute Walk Distance (6 MWD) after portopulmonary hypertension diagnosis.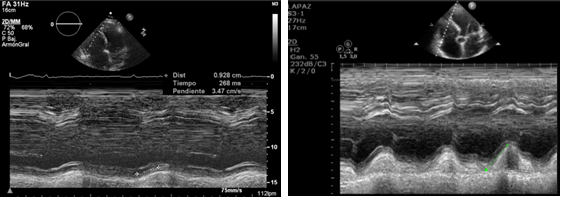 Figure 3: TAPSE (Tricuspid Annular Plane Systolic Excursion) A) At portopulmonary hypertension diagnosis; B) After treatment.
Figure 3: TAPSE (Tricuspid Annular Plane Systolic Excursion) A) At portopulmonary hypertension diagnosis; B) After treatment.One year after the start of the combination, the patient remains fully recovered, presenting WHO functional class I with no dyspnoea and no limitations in his daily activities.
DISCUSSION
To the best of our knowledge, this is the first case reported of portopulmonary hypertension treated with macitentan in combination with tadalafil. Recently, a similar clinical case was reported by Vionnet et al. [7], successfully managing a 37-year-old female with decompensated cirrhosis due to autoimmune hepatitis that was diagnosed with severe POPH (Mean Pulmonary Arterial Pressure [mPAP]=93 mmHg) when referred to liver transplantation evaluation. After two months treated with macitentan (10 mg/day) plus sildenafil (20 mg three times a day), her mPAP decreased dramatically (39 mmHg), allowing liver transplantation. The patient remains well (mPAP=18 mmHg) after 9 months on macitentan 10 mg/day.
These cases, showing swift and remarkable clinical and haemodynamic improvements, suggest that macitentan may have a key role in the treatment of POPH. The efficacy and safety of this novel dual endothelin receptor antagonist has been demonstrated in clinical trials, positioning it as an important drug in the treatment of PAH [8,9]. Additionally, its liver safety profile was demonstrated in the pivotal trial SERAPHIN [6], with low incidence of elevations in liver enzymes. Similar figures of patients with ALT or AST above 3 times the upper limit of normality were observed across placebo (4.5%) and macitentan (3.4%). Additionally, the rates of peripheral oedema were also similar in both groups (18.1 vs 18.2%). Aligned with this, in our case the incidence of peripheral oedema was successfully solved after macitentan onset. Unfortunately, most clinical trials in PAH with specific oral therapy exclude patients with portopulmonary hypertension. Only one post-hoc, exploratory analysis of two randomized clinical trials (PATENT-1/-2) including 11 patients with POPH treated with riociguat, a soluble guanylate cyclase stimulator, has been published to date [10]. After 12 weeks of treatment, median 6 MWD increased from baseline by 48 m, and 4 patients improved their WHO functional class (PATENT-1). These improvements were maintained after 2 years in the open label extension (PATENT-2). Additionally, the efficacy and safety of ambrisentan has been tested in a prospective, open-label trial (ANGEL) including 31 patients with POPH [11]. Significant improvements in mPAP and cardiac output were reported after 24 weeks of treatment, and there was a trend to an improvement in 6 MWD and NYHA functional class, although statistically significant effects were not observed.
Besides this, the availability of related data is limited to retrospective studies [12] or short case series that evaluate the efficacy of bosentan or ambrisentan, taking as a prognostic variable the 6 MWT, and with no haemodynamic follow up in most cases [13]. To date, the most effective, evidence-based treatments, particularly for patients on the waiting list for liver transplantation, are the intravenous prostanoids [14].
A randomized, double blind, placebo controlled clinical trial of macitentan in patients with POPH is currently ongoing (PORTICO trial). The main aim of this study is to evaluate the effect of macitentan on pulmonary vascular resistance after 12 weeks of treatment. 84 patients have been recruited [15], constituting the largest randomized clinical trial in patients with POPH. The results of this trial are highly awaited, since it will provide relevant information regarding the treatment of this subgroup of patients, whose particularly poor prognosis would be greatly improved with the addition of a new efficacious and safe therapy.
These cases, showing swift and remarkable clinical and haemodynamic improvements, suggest that macitentan may have a key role in the treatment of POPH. The efficacy and safety of this novel dual endothelin receptor antagonist has been demonstrated in clinical trials, positioning it as an important drug in the treatment of PAH [8,9]. Additionally, its liver safety profile was demonstrated in the pivotal trial SERAPHIN [6], with low incidence of elevations in liver enzymes. Similar figures of patients with ALT or AST above 3 times the upper limit of normality were observed across placebo (4.5%) and macitentan (3.4%). Additionally, the rates of peripheral oedema were also similar in both groups (18.1 vs 18.2%). Aligned with this, in our case the incidence of peripheral oedema was successfully solved after macitentan onset. Unfortunately, most clinical trials in PAH with specific oral therapy exclude patients with portopulmonary hypertension. Only one post-hoc, exploratory analysis of two randomized clinical trials (PATENT-1/-2) including 11 patients with POPH treated with riociguat, a soluble guanylate cyclase stimulator, has been published to date [10]. After 12 weeks of treatment, median 6 MWD increased from baseline by 48 m, and 4 patients improved their WHO functional class (PATENT-1). These improvements were maintained after 2 years in the open label extension (PATENT-2). Additionally, the efficacy and safety of ambrisentan has been tested in a prospective, open-label trial (ANGEL) including 31 patients with POPH [11]. Significant improvements in mPAP and cardiac output were reported after 24 weeks of treatment, and there was a trend to an improvement in 6 MWD and NYHA functional class, although statistically significant effects were not observed.
Besides this, the availability of related data is limited to retrospective studies [12] or short case series that evaluate the efficacy of bosentan or ambrisentan, taking as a prognostic variable the 6 MWT, and with no haemodynamic follow up in most cases [13]. To date, the most effective, evidence-based treatments, particularly for patients on the waiting list for liver transplantation, are the intravenous prostanoids [14].
A randomized, double blind, placebo controlled clinical trial of macitentan in patients with POPH is currently ongoing (PORTICO trial). The main aim of this study is to evaluate the effect of macitentan on pulmonary vascular resistance after 12 weeks of treatment. 84 patients have been recruited [15], constituting the largest randomized clinical trial in patients with POPH. The results of this trial are highly awaited, since it will provide relevant information regarding the treatment of this subgroup of patients, whose particularly poor prognosis would be greatly improved with the addition of a new efficacious and safe therapy.
ACKNOWLEDGEMENT
Medical writing assistance: Juan Martín Alonso (TFS S.L.), funded by Actelion Spain.
Actelion did not have any role in the collection, analysis and interpretation of data; writing the report; and the decision to submit the report for publication. The views expressed are therefore based on author’s opinions and do not represent the views of Actelion.
Actelion did not have any role in the collection, analysis and interpretation of data; writing the report; and the decision to submit the report for publication. The views expressed are therefore based on author’s opinions and do not represent the views of Actelion.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- Hadengue A, Benhayoun MK, Lebrec D, Benhamou JP (1991) Pulmonary hypertension complicating portal hypertension: Prevalence and relation to splanchnic hemodynamics. Gastroenterology 100: 520-528.
- Colle IO, Moreau R, Godinho E, Belghiti J, Ettori F, et al. (2003) Diagnosis of portopulmonary hypertension in candidates for liver transplantation: A prospective study. Hepatology 37: 401-409.
- Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, et al. (2013) Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 62: 34-41.
- DuBrock HM, Channick RN, Krowka MJ (2015) What’s new in the treatment of portopulmonary hypertension? Expert Rev Gastroenterol Hepatol 9: 983-992.
- Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, et al. (2015) Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. The New England Journal of Medicine 373: 834-844.5. Galiè N, Barberà JA, Frost AE, Ghofrani HA, Hoeper MM, et al. (2015) Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. The New England Journal of Medicine 373: 834-844.
- Pulido T, Adzerikho I, Channick RN, Delcroix M, Galiè N, et al. (2013) Macitentan and morbidity and mortality in pulmonary arterial hypertension. N Engl J Med 369: 809-818.
- Vionnet J, Yerly P, Aubert JD, Pascual M, Aldenkortt F, et al. (2018) Management of severe portopulmonary hypertension with dual oral therapy before liver transplantation. Transplantation 102: 194.
- Keating GM (2016) Macitentan: A review in pulmonary arterial hypertension. Am J Cardiovasc Drugs 16: 453-460.
- Dingemanse J, Sidharta PN, Maddrey WC, Rubin LJ, Mickail H (2014) Efficacy, safety and clinical pharmacology of macitentan in comparison to other endothelin receptor antagonists in the treatment of pulmonary arterial hypertension. Expert Opin Drug Saf 13: 391-405.
- Cartin Ceba R, Halank M, Ghofrani HA, Humbert M, Mattson J, et al. (2018) Riociguat treatment for portopulmonary hypertension: A subgroup analysis from the PATENT-1/-2 studies. Pulm Circ 8.
- Preston IR, Burger CD, Battarjee WF, Bartolome S, Safdar Z, et al. (2017) Ambrisentan in portopulmonary hypertension, results from the multicenter prospective ANGEL study (Ambrisentan usage in portopulmonary hypertension). American Journal of Respiratory and Critical Care Medicine 195: 6908.
- Savale L, Magnier R, Le Pavec J, Jaïs X, Montani D, et al. (2013) Efficacy, safety and pharmacokinetics of bosentan in portopulmonary hypertension. Eur Respir J 41: 96-103.
- Halank M, Knudsen L, Seyfarth HJ, Ewert R, Wiedemann B, et al. (2011) Ambrisentan improves exercise capacity and symptoms in patients with portopulmonary hypertension. Z Gastroenterol 49: 1258-1262.
- Raevens S, De Pauw M, Reyntjens K, Geerts A, Verhelst X, et al. (2013) Oral vasodilator therapy in patients with moderate to severe portopulmonary hypertension as a bridge to liver transplantation. Eur J Gastroenterol Hepatol 25: 495-502.
- NIH (2017) Portopulmonary hypertension treatment with macitentan - a randomized clinical trial (PORTICO) [NCT02382016]. NIH, Maryland, USA.
SUPPLEMENTARY FILE
SUPPLEMENTARY FILE
Citation: Soto Abánades CI, Alcolea Batres S, Sánchez Recalde A, Guzmán Martínez G, Fernández-Velilla Peña M, et al., (2018) Clinical and Haemodynamic Outcomes of the Macitentan Plus Tadalafil Combination for Severe Portopulmonary Hypertension: First Experience in Routine Clinical Practice. J Pulm Med Respir Res 4: 017.
Copyright: © 2018 Clara Itzíar Soto Abánades, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!