Journal of Pulmonary Medicine & Respiratory Research Category: Medical
Type: Case Report
Complex Tuberous Sclerosis Associated with Pulmonary Lymphangioleiomyomatosis
*Corresponding Author(s):
Dal Maso ViniciusDepartment Of Pulmonary Medicine, Universidade Federal Fronteira Sul, Passo Fundo, Rio Grande Do Sul, Brazil
Tel: +55 54996411005,
Email:vinibuaesdalmaso@yahoo.com.br
Received Date: Dec 29, 2018
Accepted Date: Jan 16, 2019
Published Date: Jan 30, 2019
Abstract
Complex tuberous sclerosis is an autosomal dominant, sporadic, or familial neurocutaneous disease that affects multiple organs and systems, commonly leads to lymphangioleiomyomatosis and benign tumors in various tissues, possibly evolving with organic dysfunction. We present a case of complex tuberous sclerosis associated with lymphangioleiomyomatosis, whose initial presentation was bilateral pneumothorax, attended at the São Vicente de Paulo Hospital, Passo Fundo, and RS.
Keywords
Bilateral pneumothorax; Case reports; Lymphangioleiomyomatosis; Tuberous sclerosis
INTRODUCTION
Complex tuberous sclerosis is a hereditary neurocutaneous disease with pleomorphic features involving multiple organs and systems [1]. The most common manifestations are benign tumors in the skin, brain, kidneys, lungs and eyes, and may lead to organ dysfunction due to parenchymal replacement by the tumors [2]. It is an autosomal dominant disease [3], with benign characteristic and phenotypic presentations of variable intensity [4], with challenging management, besides the treatment of clinical support, the use of mTOR is possible [5]. Lymphangioleiomyomatosis (LAM) is a multisystem disorder that primarily affects the lung sporadically (sporadic-LAM) or in association with TSC (TSC-LAM).
CASE REPORT
A 44-year-old female patient admitted to the emergency room for bilateral pneumothorax. History of dry cough and dyspnea on physical exertion, progressive with sudden worsening two days before hospital admission.
The patient was presented in a regular state, disoriented in time and space with blood pressure 140/100 mmhg heart rate 128 bpm, respiratory rate 28 bpm, peripheral O2 saturation of 92% with O2 at 9 l/min, ventilatory effort, pulmonary auscultation with bilaterally reduced vesicular murmur and presence of subcutaneous emphysema in the abdominal region. The patient presented lesions in the malar region of the face, called angiofibromas and fibromas in the nail bed (Figure 1).
The patient was presented in a regular state, disoriented in time and space with blood pressure 140/100 mmhg heart rate 128 bpm, respiratory rate 28 bpm, peripheral O2 saturation of 92% with O2 at 9 l/min, ventilatory effort, pulmonary auscultation with bilaterally reduced vesicular murmur and presence of subcutaneous emphysema in the abdominal region. The patient presented lesions in the malar region of the face, called angiofibromas and fibromas in the nail bed (Figure 1).
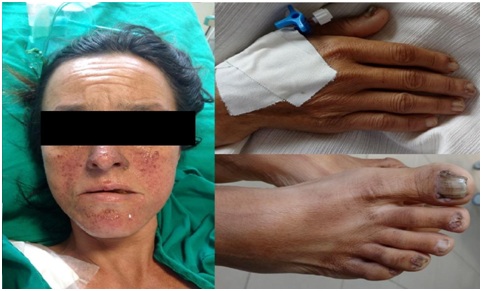 Figure 1: Angiofibromas in the malar region and fibromas in the nail bed.
Figure 1: Angiofibromas in the malar region and fibromas in the nail bed.Computed tomography of the chest reveals bilateral pneumothorax, pulmonary parenchyma with multiple cysts and area of consolidation in the left lower lobe (Figure 2). Laboratory tests of arrival with loss of renal function, creatinine 2.18, urea 86, hemoglobin 11.4, normocytic and normochromic anemia, left-sided leukocytosis, leukocytes 38400 (band cells 1920, segmented 32640, lymphocytes 1920, monocytes 1920), 480,000 platelets, serology for HIV, hepatitis B and C, toxoplasmosis, cytomegalovirus and non-reactive VDRL.
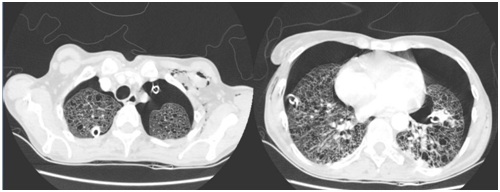 Figure 2: Computed tomography of the chest showing bilateral pneumothorax, presence of bilateral chest drain, multiple cysts in the pulmonary parenchyma, subcutaneous emphysema and consolidation in the left lower lobe.
Figure 2: Computed tomography of the chest showing bilateral pneumothorax, presence of bilateral chest drain, multiple cysts in the pulmonary parenchyma, subcutaneous emphysema and consolidation in the left lower lobe.In the management of the patient, thoracic drainage was performed in continuous aspiration, antibiotic therapy for community acquired pneumonia, supplemental oxygen, hydration, diuresis control, nasal probing for nutrition and prophylaxis for venous thromboembolism.
In previous morbid history, there are convulsive seizures with no definite diagnosis associated with cognitive deficit and learning difficulties since childhood. Electroencephalogram that detected generalized slow outbreaks in the right and left temporal lobe and magnetic resonance of the brain (Figure 3) with periventricular ependymal nodules and cortical tufts. Diagnosis of cognitive deficit and epilepsy.
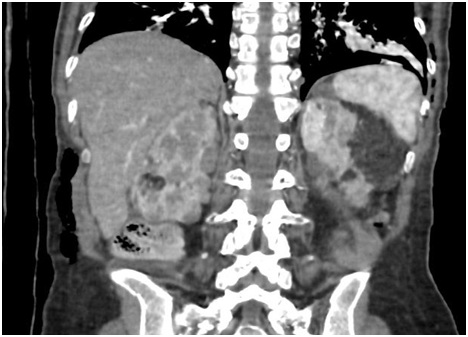 Figure 3: Computed tomography of the abdomen showing renal angiomyolipomas.
Figure 3: Computed tomography of the abdomen showing renal angiomyolipomas.Echocardiogram with ejection fraction 67.3%, left ventricular function preserved, mid degree mitral and tricuspid regurgitation and possible pulmonary hypertension. In the tomography of abdomen evidenced renal angiomyolipomas (Figure 4).
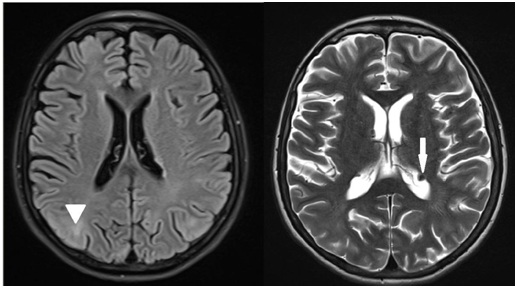 Figure 4: Magnetic resonance imaging of encephalus showing cortical tufts (arrowhead) and periventricular subependymal nodules (arrow).
Figure 4: Magnetic resonance imaging of encephalus showing cortical tufts (arrowhead) and periventricular subependymal nodules (arrow).In the patient's evolution, after a few days of hospitalization the chest drains were removed from the aspiration, but persisted with air leakage, so chemical pleurodesis with talc was performed. Talcum, on the other hand, was used since lung transplantation was not planned due to the difficulty in performing it in Brazil and also because of the poor family condition, which would certainly contraindicate the procedure.
Patient had a satisfactory evolution and was discharged, with outpatient return orientation. Due to the low socioeconomic level, patient lost the follow-up in the institution and genetic evaluation was not achieved. The mTOR inhibitors therapy was not started for the same reason.
DISCUSSION
Lymphangioleiomyomatosis (LAM) is a disease with highly variable phenotypic expression, on the initial age, disease severity and clinical manifestations [4]. It is an autosomal dominant disease [3]. Caused by a mutation in the TSC1 or TSC2 gene, mutations in TSC2 are four times more common, but in the familiar cases the prevalence of both are similar [6].
The TSC1 gene, located on chromosome 9q34, encodes hamartin and TSC2 on chromosome 16p13, encodes tuberculin. It is now recognized that the primary function of the TSC1-TSC2 complex is as a critical negative regulator of mTORC1 activation. Furthermore, the resulting elevation in mTORC1 activity is essential for tumour development [7]. Sirolimus, current therapy, inhibits mTOR activity and causes apoptosis in renal tumors [8].
LAM has benign appearance, but it is a metastatic disease from a single source and as yet unidentified, the cells of all lesions have a similar appearance, suggesting a common source [2]. Diagnosis and management is challenging, with diverse manifestations and variables and different degrees of severity [5]. We present the case of a 44-year-old patient with multiple organ manifestations, but with mild symptoms that never motivated the search for the health system.
The diagnosis is established using clinical criteria or genetic testing, and the identification of the mutation in genes TSC1 and TSC2 in an uninjured tissue is sufficient for diagnosis, if the patient does not present clinical manifestation, but 10-25% of the affected patients do not present the mutation [2]. The initial presentation we did not perform a genetic test because the patient had multiple clinical manifestations and the genetic mapping has difficult access with high costs.
Skin manifestations present are facial angiofibromas, more common in the malar region. Ungual fibroids, hypomelanocitic macules, and Shagreen marks that are large plaques on the back, with uneven relief or appearance of orange peel [2]. Among the typical characteristics, the only one not presented by the patient is the Shagren's mark.
Associated neuropsychiatric disorders cause great morbidity and mortality [2], generating autistic spectrum disorders, attention deficit disorder and hyperactivity, severe epilepsy, and mild to profound intellectual disability [5]. Cerebral tumors associated are cortical tubercules, subependymal nodules and subependymal giant cell astrocytoma [2]. The patient has subependymal nodules and cortical tubercules, expected findings in the spectrum of the disease, as well as epilepsy and mental retardation, signs neglected by relatives, because they have been present since childhood.
The most common lung lesion is lymphangioleiomyomatosis, characterized by cystic destruction of the parenchyma, leading to progressive decline in lung function. Patients present dyspnea on exertion and spontaneous pneumothorax resulting from subpleural vesicle rupture and may be the first manifestation of the disease. Another possible manifestation is chylous pulmonary congestion, a result of lymphatic obstruction by muscle cells [8]. Chest X-rays show nonspecific alterations, whereas diffuse, bilateral, thin-walled cysts are described in chest tomography, with a size between 1 and 45mm diameter, varies from small to even complete replacement of the normal parenchyma. There may be hilar or mediastinal lymphadenopathy [8]. The definitive diagnosis is with pulmonary biopsy, with HMB-45 staining, which is highly specific for LAM. Transbronchial biopsy can also be performed, with yield up to 60%, but the procedure guided by thoracoscopy the gold standard, is hardly indicated [8].
The fact that motivated the investigation of the clinical picture of the present report was the presence of bilateral spontaneous pneumothorax, being guided by cystic substitution of the pulmonary parenchyma on the chest tomography. No pulmonary biopsy was performed, as previously mentioned, since these are typical clinical manifestations of the disease.
The most common renal manifestations are angiomyolipomas, or multiple renal cysts. Renal impairment is an important source of morbidity and mortality, including the need for renal replacement therapy and transplantation. Angiomyolipomas are benign tumors composed of vessels, smooth muscle cells and adipose tissue, most commonly present in the kidney, but may injure other organs and cause bleeding due to its vascular nature [2]. The patient has renal angiomyolipomas, with loss of function, but without indication of substitutive therapy.
The specific treatment for lymphangioleiomyomatosis is inhibitors of the mTOR gene [5]. The currently recommended mTOR inhibitor is sirolimus, which silences phosphorylation and induces apoptosis, necrosis and regression of renal angiomyolipomas, and restores ordered cell growth [8].
For the patient with neurological manifestations, there is insufficient evidence to support their use, with individualized management for each patient [9]. In the patient with renal angiomyolipoma greater than 3 cm in diameter treatment with an mTOR inhibitor is recommended as first-line therapy for patients with neurological manifestations effective in the short term, the benefit being greater than treatment risk. Control of blood pressure and renal function is required [9].
The MILES study (Multicenter International LAM Efficacy of Sirolimus) was a double-blind, randomized, parallel group trial in which 89 patients with LAM and moderate lung impairment (defined as FEV1<70% predicted) were randomly assigned to receive treatment with sirolimus or placebo for 12 months, followed by 12 months of observation [10]. The treatment resulted in stabilization of lung function decline compared with placebo, improve FVC and quality of life measured by the EuroQOL scale. The most common adverse events were mucositis, diarrhea, nausea, hypercholesterolemia, acneiform rash, and swelling in the lower extremities.
For moderate or severe pulmonary disease or on rapid progression mTOR inhibitor treatment may be used to stabilize or improve lung function, as well as quality of life and functional performance [9]. Sirolimus is recommended if FEV1 is below of 70% of predicted or for chylous collections. It is important to recommend pulmonary rehabilitation and use of supplemental oxygen to maintain peripheral oxyhemoglobin saturation above 90%. There is no recommendation for pleural sclerosis to prevent new pneumothoraxes. Lung transplantation is indicated if FEV1 is lower than 30% of predicted [4]. In the patient in question, pleurodesis with talc was performed due to the difficulty of removing the chest drain due to the constant air leakage through the drains. No specific therapy with sirolimus was initiated during hospitalization due to the low cognitive level of the patient and the family, early ambulatory follow-up was proposed for clinical evaluation and initiation of optimized treatment. The patient returned to the outpatient appointment, but did not perform the requested exams, reinforcing the therapeutic limitation for patients with low cognitive, socioeconomic and low adherence to the proposed recommendations.
The TSC1 gene, located on chromosome 9q34, encodes hamartin and TSC2 on chromosome 16p13, encodes tuberculin. It is now recognized that the primary function of the TSC1-TSC2 complex is as a critical negative regulator of mTORC1 activation. Furthermore, the resulting elevation in mTORC1 activity is essential for tumour development [7]. Sirolimus, current therapy, inhibits mTOR activity and causes apoptosis in renal tumors [8].
LAM has benign appearance, but it is a metastatic disease from a single source and as yet unidentified, the cells of all lesions have a similar appearance, suggesting a common source [2]. Diagnosis and management is challenging, with diverse manifestations and variables and different degrees of severity [5]. We present the case of a 44-year-old patient with multiple organ manifestations, but with mild symptoms that never motivated the search for the health system.
The diagnosis is established using clinical criteria or genetic testing, and the identification of the mutation in genes TSC1 and TSC2 in an uninjured tissue is sufficient for diagnosis, if the patient does not present clinical manifestation, but 10-25% of the affected patients do not present the mutation [2]. The initial presentation we did not perform a genetic test because the patient had multiple clinical manifestations and the genetic mapping has difficult access with high costs.
Skin manifestations present are facial angiofibromas, more common in the malar region. Ungual fibroids, hypomelanocitic macules, and Shagreen marks that are large plaques on the back, with uneven relief or appearance of orange peel [2]. Among the typical characteristics, the only one not presented by the patient is the Shagren's mark.
Associated neuropsychiatric disorders cause great morbidity and mortality [2], generating autistic spectrum disorders, attention deficit disorder and hyperactivity, severe epilepsy, and mild to profound intellectual disability [5]. Cerebral tumors associated are cortical tubercules, subependymal nodules and subependymal giant cell astrocytoma [2]. The patient has subependymal nodules and cortical tubercules, expected findings in the spectrum of the disease, as well as epilepsy and mental retardation, signs neglected by relatives, because they have been present since childhood.
The most common lung lesion is lymphangioleiomyomatosis, characterized by cystic destruction of the parenchyma, leading to progressive decline in lung function. Patients present dyspnea on exertion and spontaneous pneumothorax resulting from subpleural vesicle rupture and may be the first manifestation of the disease. Another possible manifestation is chylous pulmonary congestion, a result of lymphatic obstruction by muscle cells [8]. Chest X-rays show nonspecific alterations, whereas diffuse, bilateral, thin-walled cysts are described in chest tomography, with a size between 1 and 45mm diameter, varies from small to even complete replacement of the normal parenchyma. There may be hilar or mediastinal lymphadenopathy [8]. The definitive diagnosis is with pulmonary biopsy, with HMB-45 staining, which is highly specific for LAM. Transbronchial biopsy can also be performed, with yield up to 60%, but the procedure guided by thoracoscopy the gold standard, is hardly indicated [8].
The fact that motivated the investigation of the clinical picture of the present report was the presence of bilateral spontaneous pneumothorax, being guided by cystic substitution of the pulmonary parenchyma on the chest tomography. No pulmonary biopsy was performed, as previously mentioned, since these are typical clinical manifestations of the disease.
The most common renal manifestations are angiomyolipomas, or multiple renal cysts. Renal impairment is an important source of morbidity and mortality, including the need for renal replacement therapy and transplantation. Angiomyolipomas are benign tumors composed of vessels, smooth muscle cells and adipose tissue, most commonly present in the kidney, but may injure other organs and cause bleeding due to its vascular nature [2]. The patient has renal angiomyolipomas, with loss of function, but without indication of substitutive therapy.
The specific treatment for lymphangioleiomyomatosis is inhibitors of the mTOR gene [5]. The currently recommended mTOR inhibitor is sirolimus, which silences phosphorylation and induces apoptosis, necrosis and regression of renal angiomyolipomas, and restores ordered cell growth [8].
For the patient with neurological manifestations, there is insufficient evidence to support their use, with individualized management for each patient [9]. In the patient with renal angiomyolipoma greater than 3 cm in diameter treatment with an mTOR inhibitor is recommended as first-line therapy for patients with neurological manifestations effective in the short term, the benefit being greater than treatment risk. Control of blood pressure and renal function is required [9].
The MILES study (Multicenter International LAM Efficacy of Sirolimus) was a double-blind, randomized, parallel group trial in which 89 patients with LAM and moderate lung impairment (defined as FEV1<70% predicted) were randomly assigned to receive treatment with sirolimus or placebo for 12 months, followed by 12 months of observation [10]. The treatment resulted in stabilization of lung function decline compared with placebo, improve FVC and quality of life measured by the EuroQOL scale. The most common adverse events were mucositis, diarrhea, nausea, hypercholesterolemia, acneiform rash, and swelling in the lower extremities.
For moderate or severe pulmonary disease or on rapid progression mTOR inhibitor treatment may be used to stabilize or improve lung function, as well as quality of life and functional performance [9]. Sirolimus is recommended if FEV1 is below of 70% of predicted or for chylous collections. It is important to recommend pulmonary rehabilitation and use of supplemental oxygen to maintain peripheral oxyhemoglobin saturation above 90%. There is no recommendation for pleural sclerosis to prevent new pneumothoraxes. Lung transplantation is indicated if FEV1 is lower than 30% of predicted [4]. In the patient in question, pleurodesis with talc was performed due to the difficulty of removing the chest drain due to the constant air leakage through the drains. No specific therapy with sirolimus was initiated during hospitalization due to the low cognitive level of the patient and the family, early ambulatory follow-up was proposed for clinical evaluation and initiation of optimized treatment. The patient returned to the outpatient appointment, but did not perform the requested exams, reinforcing the therapeutic limitation for patients with low cognitive, socioeconomic and low adherence to the proposed recommendations.
REFERENCES
- Gokare P, El-Deiry WS (2017) The tuberosis sclerosis complex gets fatter. Oncotarget 8: 41780-41781.
- Northrup H, Krueger DA, International Tuberous Sclerosis Complex Consensus Group (2013) Tuberous sclerosis complex diagnostic criteria update: Recommendations of the 2012 Iinternational Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 49: 243-254.
- Curatolo P, Bombardieri R, Jozwiak S (2008) Tuberous sclerosis. Lancet 372: 657-668.
- Smalley SL, Burger F, Smith M (1994) Phenotypic variation of tuberous sclerosis in a single extended kindred. J Med Genet 31: 761-765.
- Johnson MP, Johnson JC, Engel-Nitz NM, Said Q, Prestifilippo J, et al. (2017) Management of a rare disease population: A model for identifying a patient population with tuberous sclerosis complex. Management of Tuberous Sclerosis Complex.
- Au KS, Williams AT, Roach ES, Batchelor L, Sparagana SP, et al. (2007) Genotype/phenotype correlation in 325 individuals referred for a diagnosis of tuberous sclerosis complex in the United States. Genet Med 9: 88-100.
- Huang J, Manning BD (2008) The TSC1-TSC2 complex: A molecular switchboard controlling cell growth. Biochem J 412: 179-190.
- Broaddus VC, Mason RJ, Ernst JD, King TE, Lazarus SC, et al. (2017) Murray & Nadel tratado de medicina respiratoria. Elsevier Brasil, Rio de Janeiro, Brazil. Pg no: 1243-1259.
- Krueger DA, Northrup H (2013) Tuberous sclerosis complex surveillance and management: Recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatr Neurol 49: 255-265.
- McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, et al. (2011) Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 364: 1595-1606.
Copyright: © 2019 Bogoni Giuliane, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!