Cystic Fibrosis in India: Past, Present and Future
*Corresponding Author(s):
Sushil K KabraDepartment Of Pediatrics, All India Institute Of Medical Sciences, New Delhi 110029, India
Tel:+91 1126594610,
Email:skkabra@hotmail.com
Abstract
Keywords
Airway clearance technique; CFTR mutation; Cystic fibrosis; Delayed diagnosis; Developing countries; India; Malnutrition; Pancreatic enzyme replacement therapy; Sweat test
INTRODUCTION
The 1950s saw the beginning of the sweat test developed as a result of discoveries made by Paul di Sant’Agnese during the heat wave in New York in 1953 [3]. It was later standardized by Gibson and Cooke in 1959 [4]. In 1955, Cystic Fibrosis Foundation was founded and chloride transport was identified as the basic physiologic defect of CF in 1983. In 1985 gene causing cystic fibrosis was narrowed down to chromosome 7. This paved the way for Professor Lap-Chi Tsui and colleagues to identify the specific faulty (cystic fibrosis transmembrane conductance regulator) gene in 1989. More recent publication though suggests that it is not CFTR, rather the Proteostasis Network (PN)/and ‘Social Network’ (SN) and their impact on the CFTR Functional Landscape (CFFL) are the key mechanisms that can explain clinical features and progression of illness [5]. Better understanding of these processes may change the approach to treatment of CF in future.
Cystic Fibrosis (CF), an autosomal recessive monogenic disorder, is the commonest inherited life limiting illness initially considered to be affecting the Caucasians only. Over past few decades it has been recognized that CF exists in all ethnic groups. CF is a multisystem disorder characterized by involvement of lungs, pancreas and other exocrine glands, manifesting with recurrent respiratory infections, malabsorption and a myriad of complications pertaining to other systems, namely hepatobilliary, endocrine and reproductive. The dominant morbidity remains secondary to pulmonary involvement with commonest cause of mortality being respiratory failure following steady decline in lung function. The role of nutrition on lung function and subsequent outcome in children from India, a developing country with huge burden of under nutrition can never be overemphasized. Ong et al., in their study on Indian children [6] suggested that nutritional differences influence qualitative aspects of lung development in childhood beyond simple isotropic lung growth. They also predicted that lung function must take account of these differences if disease related changes are to be accurately measured.
Over past 5-6 decades with better understanding of pathogenesis of CF, significant improvement has occurred in treatment manifesting as survival benefit. The Cystic Fibrosis Foundation (CFF) has projected a life expectancy of 37 years for CF patients currently [7] and a UK model predicts that a child born with CF today can expect to live past 50 years of age [8]. In countries with limited resources like India, the survival of children with CF is improving but lags considerably behind the developed countries. Early diagnosis and institution of appropriate treatment is the key to improve survival in children with CF. It is important to create awareness among pediatricians of developing countries. It can be achieved by reviewing information about CF in individual countries and removing myth that it is disease of Caucasian population alone. Still much has changed in the understanding, identification and management of cystic fibrosis in this country. The main aim of this article is to review details of CF including magnitude of problem, clinical manifestations, how to suspect and diagnose and how to manage suspected children with CF with limitation of resources.
MAGNITUDE OF PROBLEM
Cystic fibrosis, once thought to be non-existent in India, was first reported in 1968 [12,13]. Thereafter, there have been reports of CF from all parts of India (Table 1), suggesting that CF occurs in Indian population possibly throughout the geographic or ethnic groups. Literature search suggest a rapid rise of publications related to CF in India over past two decades. A MEDLINE search using CF and India as search words shows increasing citations with time: for the period 1968-1970: 5 citations, 1971-1980: only two citations, 1981-1990: 5 citations, 1991-2000: 22 citations, 2001-2010: 74 citations and from 2011-2015 (December): 117 citations.
| Author, year of publication (Ref no) | Number of cases | Diagnostic method used |
| Bhakoo et al., 1968 [12] | 1 | Histopathology |
| Mehta et al., 1968 [13] | 13 | Sweat test |
| Mehta et al., 1969 [14] | 6 | Sweat test |
| Devi et al., 1969 [15] | 4 | Sweat test |
| Gupte et al., 1970 [16] | 6 | Sweat test |
| Reddy et al., 1970 [17] | 12 | Autopsy |
| Venkataraman et al., 1972 [18] | 1 | Autopsy |
| Maya PP et al., 1980 [19] | 3 | Sweat test |
| Jagdish JS 1989 [20] | 1 | Sweat test |
| Prasad ML et al.1990 [21] | 2 | Autopsy |
| Deivanayagam et al., 1990 [22] | 5 | Sweat test |
| Sarkar AK et al., 1992 [23] | 1 | Sweat test |
| Kabra et al., 1996 [24] | 15 | Sweat test, Mutations |
| Kabra et al., 1996 [25] | 13 | Sweat test |
| Kabra et al., 1999 [26] | 62 | Sweat test, Mutations |
| Kabra et al., 2000 [27] | 24 | Sweat test, Mutations |
| Singh M et al., 2002 [28] | 17 | Sweat test, Mutation, Autopsy |
| Kabra et al., 2003 [29] | 120 | Sweat test, Mutations |
| Ashavaid et al., 2003 [30] | 5 | Sweat test, Mutations |
| Ashavaid et al., 2005 [31] | 39 | Sweat test, Mutations |
| Sharma N et al., 2009 [32] | 50 | Sweat test, Mutations, Autopsy, IRT* |
| Mir TA et al., 2011 [33] | 3 | Sweat test, Mutations |
| Santra G et al., 2012 [34] | 1 | Sweat test |
| Kawoosa MS et al., 2013 [35] | 18 | Sweat test, Mutations |
| Chakraborty et al., 2014 [36] | 2 | Sweat test |
| Sharma et al., 2015 [37] | 1 | Autopsy |
There are no community based studies to document precise incidence/prevalence of CF in India. An estimation based on number of CF children registered in CF clinics from Indian subcontinent and total population in defined geographic region has reported 1:12000 from UK and 1:40000 from US [38,39]. A study on 950 cord blood samples investigating carrier state of ΔF508 mutation from India calculated incidence of CF as 1:40000 newborns [40]. Available literature suggests that the prevalence of CF and ΔF508 mutation is far more common in northern part of the country compared to South India [29]. Similarly, in Pakistan, ΔF508 mutation is most frequently reported from the Baluchistan region (South Eastern Pakistan). Baluchistan was hypothesized to be the cradle of this particular mutation from where population migration might have carried the same to India [41].
ESTIMATED BURDEN OF CF IN INDIA
Genetics and molecular defect
There are only few studies that describe genotypes of Indian children with CF. The frequency of ΔF508 mutation in Indian children with CF has been reported between 19 to 56% [46]. Also in other Asian countries, the reported proportion of children with ΔF508 is less than that seen in Caucasian population [47]. The spectrum of mutations apart from ΔF508 in Indian patients is highly heterozygous and some rare and new mutations are also described [27] (Table 2). Detailed analysis suggests that there is variation in mutation profile of children with CF [48]. Children born to parents who had their ancestry from Pakistan had frequency of ΔF508 almost similar to that of Caucasian population [29]. The heterogeneity in mutation profile is possibly due to variation in ethnic background.
| Authors [Reference] | Total number of patients | Percentage of patients In whom mutation identified | Mutations identified |
| Kabra et al., [25] | 13 | 61 |
Homozygous ΔF508:6 Heterozygous ΔF508:2* |
| Kabra et al., [27] | 24 | 75 |
Homozygous ΔF508:5 |
| Singh et al., [28] | 17 | 25 (70% patients tested for mutation) | ΔF508* |
| Kabra et al., [29] | 120 | 22 |
Homozygous ΔF508:19 Heterozygous ΔF508:7 |
| Ashavaid et al., [30] | 23** |
Homozygous ΔF508:4 Heterozygous ΔF508:1 |
|
| Ashavaid et al., [31] | 37 | 24 |
ΔF508 (24%)*** 1525-1G > A |
| Sharma et el., [32] | 50 | 98 |
Homozygous ΔF508:5 |
| Shastri et al., [48] | 100 | 41 |
Homozygous ΔF508l:20 |
| Sachdeva et al., [49] | 225 | 35 |
Homozygous ΔF508l:45 |
| Kawoosa et al., [35] | 18 | 83% of patients were tested using mutation analysis |
Homozygous ΔF508l:3 |
Clinical manifestations
| Characteristics | Frequency (N = 120) |
| Demography | |
| Mean age at diagnosis | 54 months (95% CI 3-154) |
| Mean age of onset of symptoms | 11 months (95% CI 0.1-60) |
| Boys | 80 (67%) |
| Girls | 40 (33%) |
| Symptoms | |
| Persistent/recurrent pneumonia | 118 (98%) |
| Failure to thrive | 108 (90%) |
| Malabsorption | 96 (80%) |
| Rectal prolapse | 16 (13%) |
| Dehydration | 16 (13%) |
| Meconium ileus | 10 (9%) |
| Vitamin A deficiency | 10 (9%) |
| Salt craving | 5 (5%) |
| Salty taste | 5 (5%) |
| Skin rashes | 5 (5%) |
| Vitamin D deficiency | 4 (4%) |
| Pneumothorax/empyema | 3 (3%) |
| Meconium ileus equivalent | 2 (2%) |
| Examination | |
| Normal or mild malnutrition | 70 (58%) |
| Severe malnutrition | 50 (42%) |
| Z Scores for weight for age | - 2.59 (95% CI: - 3.01 to -2.32) |
| Clubbing | 80 (75%) |
| Chest | |
| Crepitations | 110 (92%) |
| Hyperinflation | 100 (83%) |
| Rhonchi | 40 (33%) |
| Bronchial breathing | 20 (17%) |
| Nasal polyposis | 5 (4%) |
| CF score mean (95% CI) | 51 (20-80) |
The high rate of micronutrient deficiencies in Indian children with CF is highlighted in recent studies also [50]. The frequency of colonization by Pseudomonas aeruginosa and antibiotic resistance was higher in Indian children with CF and few were colonized by phenotypically and genotypically distinct strains of the organism [51]. In a recent study from our center [52], CF patients were observed to have high prevalence of peripheral neuropathy. Prevalence of ABPA was also found to be higher in Indian children with CF [53].
In the absence of CFTR expression, fluid secretion into the gut is reduced and the contents get inspissated. In the neonatal period, this manifests as meconium ileus, and later in life, as distal intestinal obstruction syndrome. Few patients of CF, who have residual pancreatic function, can suffer from recurrent attacks of pancreatitis.
CF Related Diabetes (CFRD) affects 2% of children with CF, while the prevalence increases with age to affect 19% of adolescents with CF [54]. Counter-regulatory hormones, like glucagon, are also compromised; therefore, ketoacidosis is seldom a complication but hyperosmolar complications and late organ damage may be observed. Corticosteroids accelerate the diabetes which in turn contributes negatively to the prognosis.
CFTR gene mutation and polymorphisms have also been associated with many diseases other than CF. Indian patients with idiopathic chronic pancreatitis were observed to have strong genetic association with CFTR gene mutation [55]. Congenital Bilateral Absence of Vas Deferens (CBAVD) is one of the known manifestations of CF in men but even non-CBAVD obstructive azoospermia was found to be associated with CFTR gene mutation in India [56].
Diagnosis
| One or more characteristic clinical features |
| -or a positive newborn screening test result |
| -or a history of CF in a sibling |
| AND |
| Increased sweat chloride concentration by pilocarpine iontophoresis method on two or more occasions |
| -or identification of two CF causing mutations |
| -or demonstration of abnormal nasal epithelial ion transport |
In a child with a high pretest probability of CF, the diagnosis is confirmed by sweat chloride estimation by pilocarpine iontophoresis method. A sweat chloride concentration of > 60 mmol/L is almost always diagnostic of CF, as a falsely elevated sweat chloride in the absence of CF is rare, although reported in a number of unusual clinical conditions; they can usually be readily distinguished from CF [58]. Non-classic or atypical CF (1-2% of CF population), defines patients with a CF phenotype in at least one organ system and a normal or borderline sweat chloride level [59]. In practice, the most common cause of incorrect sweat chloride results is laboratory error; therefore, it is recommended to be done only at CFF accredited care centers [60]. But in countries like India where such facilities are not available, it should be carried out in experienced laboratories and repeated 2-3 times [61].
As already noted, CF may be diagnosed by identifying two known disease causing mutations. Using a discrete panel of mutations is faster and less costly than expanded mutational analysis. But because of heterogeneity in mutations, a single panel of mutations cannot be used for diagnosis of CF in India. Possibly multiple panel may be required for best diagnostic yield. Till that is available, sweat chloride remains gold standard for diagnosis of CF in India, though it may miss small number of children with normal sweat test having CFTR dysfunction. On the other hand, full sequence analysis will detect virtually all CFTR mutations, but its interpretation may be rather difficult as it often uncovers polymorphisms and novel mutations whose significance is not known [57].
With a huge population of around 1.25 billion, there are very few centers in India where sweat chloride test is available. Therefore, the patients have to travel long distances to get the test done and also bear the expenses for the same. So, in many case even if the diagnosis is suspected, it remains unconfirmed [62]. To overcome this problem, we developed and validated an indigenous method for sweat chloride estimation based on pilocarpine iontophoresis and manual titration [63]. The equipment was validated by doing titration of known strength of saline by two observers and by checking inter-observer variation when titration was performed on collected sweat by two independent persons. The mean difference in estimated chloride value from the known strength of saline in 50 samples was -1.04 ± 4.13 mEq/L (95% CI: -0.07 to 2.28). The mean difference in the estimated chloride values between two observers when the test was performed on known strengths of saline solution was -2.5 ± 4.24 mEq/L (95% CI: -3.67 to 1.33). The inter-observer variability between two observers when the test was performed on sweat samples obtained from 50 individuals was -1.12 ± 4.34 mEq/L (95% CI: -2.23 to 0.8).
Sweat weight of more than 100 mg could be collected in the first attempt in 602 of 757 (80%) patients with an average sweat weight of 230 mg. This inexpensive method of sweat collection and chloride estimation has acceptable accuracy and repeatability and can be used in resource poor setting for making a diagnosis of cystic fibrosis [64]. The cost of the equipment is < 10 US dollar and cost per test is < 1 US dollar.
If sweat testing facility is not available, then it as advised to look for supportive evidence for CF (Figure 1) and if they are suggestive of CF then refer the patients to a center where sweat testing facility is available.
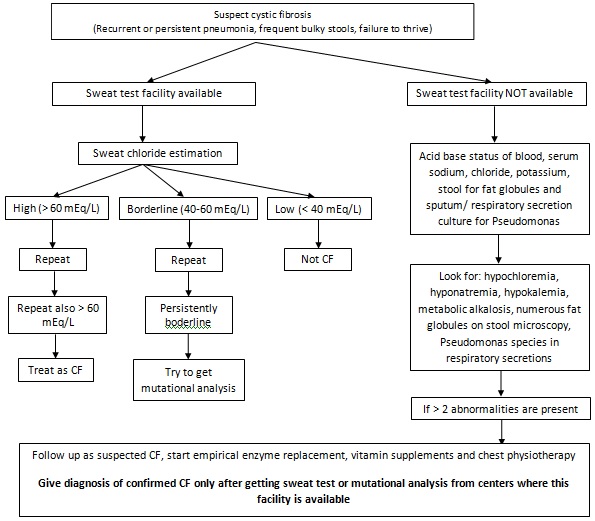
Nasal Transepithelial Potential Difference (NPD) is a labor intensive and technically difficult method available only at small numbers of CF research centers. It remains to be a research tool and has limited validation as a diagnostic test [57].
Isolation of Pseudomonas aeruginosa or Burkholdelia cepacia from sputum, cough swabs or BAL is suggestive of CF. Below 20 years of age, cystic fibrosis is the commonest cause of exocrine pancreatic insufficiency [65]. Semi quantitative estimates of fat malabsorption can be made by fecal microscopy or fecal steatocrit [66]. Recently, stool pancreatic elastase-1 has been reported as a sensitive and specific test [67].
Radiological investigations in the form of x-ray and CT scan of chest and sinuses are very non-specific. Chest imaging characteristically reveals bilateral central bronchiectasis, mucous impaction with segmental collapse, air trapping, and there is delayed pneumatization or mucosal thickening in sinuses.
MANAGEMENT
Several obstacles and hindrances are encountered by both patients and healthcare providers in management of cystic fibrosis in a developing country like India. In view of limited resources, it is very difficult to have a team of professionals from different disciplines, necessitating multitasking and heightened dedication from the available members. Availability and cost constraints for various medications (pancreatic enzymes, antibiotics, DNase, etc.) makes management of such patients very difficult. It is desirable that CF patients are followed up regularly every 4-8 weekly at a center having expertise in the management of CF. But in the Indian scenario, management of CF by individual pediatrician every 4 weeks and visit to centralized service once in 3 months may be an alternative for patients coming from far off places. Early diagnosis, regular follow up, aggressive airway clearance, use of basic microbiology and radiology do not increase cost too much but benefits are significant. Addition of pancreatic enzyme replacement and fat soluble vitamins add to cost with improvement in outcome and can be followed up in majority of patients. Addition of inhaled antibiotics and DNase are useful intervention but increase the costs significantly. Alternatives include the use of low cost treatment in form of hypertonic saline inhalation [69]. The hot and humid climate of Indian subcontinent predisposes these children for chronic dehydration and salt depletion. The scenario is further worsened by episodes of dehydration associated with gastrointestinal diseases and during summer months. The cultural practices that promote over-wrapping of infants and smaller children in Indian communities aggravate such losses. Supplementation of extra salt to these children coupled with parental education is invaluable part of their management.
Future therapies
Prognosis of CF
Factors that were found associated with decreased survival in our patients include: age at onset of symptoms < 2 months, frequency of pneumonia > 4 episodes/year, severe malnutrition at the time of diagnosis and colonization with Pseudomonas at the time of diagnosis [26]. Though we have to go a long way to match with western countries, we feel that early diagnosis, aggressive chest physiotherapy with judicious use of antibiotics and nutritional management can improve the quality of life and survival in CF patients even in developing countries like India.
Research priorities
- More number of mutation studies in Indian CF patients in an attempt to find out the full spectrum of CF mutation in this country, so that a comprehensive diagnostic panel could be formulated.
- To develop simpler ancillary tests, e.g., aquagenic palmer wrinkling [75] to aid the diagnosis.
- To formulate guidelines for daily salt supplementation in hot and humid climate like India.
- To discover newer and cheaper antibiotics, that can be given through inhaled route achieving higher lung concentrations, especially against resistant pathogens like Pseudomonas.
SUMMARY AND CONCLUSION
REFERENCES
- Cystic Fibrosis Trust (2015) History of Cystic Fibrosis. Cystic Fibrosis Trust, London, UK.
- Andersen DH (1938) Cystic fibrosis of the pancreas and its relation to celiac disease: A clinical and pathologic study. Am J Dis Child. 56: 344-399.
- Di Sant’agnese P, Darling RC, Perara GA, Shea E (1953) Abnormal electrolyte composition of sweat in cystic fibrosis of the pancreas. AMA Am J Dis Child 86: 618-619.
- Gibson LE, Cooke RE (1959) A test for concentration of electrolytes in sweat in cystic fibrosis of the pancreas utilizing pilocarpine by iontophoresis. Pediatrics 23: 545-549.
- Amaral MD, Balch WE (2015) Hallmarks of therapeutic management of the cystic fibrosis functional landscape. J Cyst Fibros 14: 687-699.
- Ong TJ, Mehta A, Ogston S, Mukhopadhyay S (1998) Prediction of lung function in the inadequately nourished. Arch Dis Child. 79: 18-21.
- Cystic Fibrosis Foundation (2008) Patient Registry, Annual Data Report 2007. Cystic Fibrosis Foundation, Bethesda, USA.
- Dodge JA, Lewis PA, Stanton M, Wilsher J (2007) Cystic fibrosis mortality and survival in the UK: 1947-2003. Eur Respir J 29: 522-526.
- Dodge JA, Morison S, Lewis PA, Coles EC, Geddes D, et al. (1997) Incidence, population, and survival of cystic fibrosis in the UK, 1968-95. UK Cystic Fibrosis Survey Management Committee. Arch Dis Child 77: 493-496.
- Hamosh A, FitzSimmons SC, Macek M, Knowles MR, Rosenstein BJ, et al. (1998) Comparison of the clinical manifestations of cystic fibrosis in black and white patients. J Pediatrics 132: 255-259.
- FitzSimmons S (1993) The changing epidemiology of cystic fibrosis. J Pediatr 122: 1-9.
- Bhakoo ON, Kumar R, Walia BNS (1968) Mucoviscidosis of lungs. Report of a case. Indian J Pediatr 35: 183-185.
- Mehta S, Wadhwa UN, Mehta SK, Chhuttani PN (1968) Fibrocystic disease of pancreas in India. Indian Pediatr 5: 185-191.
- Mehta S, Ansari Z, Wadhwa UN, Walia BN (1969) Fibrocystic disease of pancreas. Indian Pediatr 6: 114-117.
- Devi CS, Rao NR, Ramaiah Y (1969) Cystic fibrosis of pancreas in adults. A report of 4 cases. Indian J Chest Dis 11: 163-167.
- Gupte SP, Mehta S (1970) Chronic diarrhoea--an etiological study. Indian Pediatr 7: 625-627.
- Reddy CR, Sita Devi C, Anees AM, Prasantha Murthy D, Eswar Reddy G (1970) Cystic fibrosis of pancreas in India. J Trop Med Hyg 73: 59-62.
- Venkataramana G, Kousalya L, Reddy CR (1972) Cystic fibrosis of the pancreas. Indian J Pediatr 39: 337-338.
- Maya PP, Hill PG, Sudarsanam D, Jadhav M (1980) Cystic fibrosis in South India. Trop Geogr Med 32: 45-49.
- Jagadish JS (1989) Cystic fibrosis of the lungs. Indian J Pediatr 56: 288-290.
- Prasad ML, Misra A, Mathur M, Gupta DK (1990) Cystic fibrosis: postmortem report on two cases. Indian Pediatr 27: 493-496.
- Deivanayagam CN, Venogupalan K, Mallikensan S, Madhavan K, Muthukumaraswamy N (1990) A Clinical profile of cystic fibrosis in South India. Lung India 8: 167-72.
- Sarkar AK, Bag SK, Biswas SK, Saha SG (1992) Acro-osteolysis of phalanges in a case of cystic fibrosis. Indian J Pediatr 59: 636-639.
- Kabra SK, Kabra M, Ghosh M, Verma IC (1996) Cystic fibrosis--an Indian perspective on recent advances in diagnosis and management. Indian J Pediatr 63: 189-198.
- Kabra M, Ghosh M, Kabra SK, Khanna A, Verma IC (1996) Delta F508 molecular mutation in Indian children with cystic fibrosis. Indian J Med Res 104: 355-358.
- Kabra SK, Kabra M, Ghosh M, Khanna A, Pandey RM (1999) Cystic fibrosis in Indian children: clinical profile of 62 children. Pediatr Pulmonol 19: 337.
- Kabra M, Kabra SK, Ghosh M, Khanna A, Arora S, et al. (2000) Is the spectrum of mutations in Indian patients with cystic fibrosis different? Am J Med Genet 93: 161-163.
- Singh M, Prasad R, Kumar L (2002) Cystic fibrosis in North Indian children. Indian J Pediatr 69: 627-629.
- Kabra SK, Kabra M, Lodha R, Shastri S, Ghosh M, et al. (2003) Clinical profile and frequency of delta f508 mutation in Indian children with cystic fibrosis. Indian Pediatr 40: 612-619.
- Ashavaid TF, Dherai AJ, Kondkar AA, Raghavan R, Udani SV, et al. (2003) Molecular diagnosis of cystic fibrosis in Indian patients--a preliminary report. J Assoc Physicians India 51: 345-348.
- Ashavaid TF, Kondkar AA, Dherai AJ, Raghavan R, Udani SV, et al. (2005) Application of multiplex ARMS and SSCP/HD analysis in molecular diagnosis of cystic fibrosis in Indian patients. Mol Diagn 9: 59-66.
- Sharma N, Singh M, Kaur G, Thapa BR, Prasad R (2009) Identification and characterization of CFTR gene mutations in Indian CF patients. Ann Hum Genet 73: 26-33.
- Mir TA, Ashraf M, Ahmed K, Chowdhary J, Rehana B, et al. (2011) Clinical profile, diagnostic delay, and genetic make-up of cystic fibrosis in Kashmir, India. Lung India 28: 97-100.
- Santra G, Banerjee S (2012) Adult cystic fibrosis--a rare diagnosis from India. J Assoc Physicians India 60: 45-47.
- Kawoosa MS, Bhat MA, Ali SW, Hafeez I, Shastri S (2014) Clinical and mutation profile of children with cystic fibrosis in Jammu and Kashmir. Indian Pediatr 51: 185-189.
- Chakraborty PP, Ray S, Bhattacharjee R, Ghosh S, Mukhopadhyay P, et al. (2015) Diabetes and primary infertility in young males: do not forget cystic fibrosis. Clin Diabetes 33: 80-83.
- Sharma P, Arthi N, Bhattad S, Vaiphei K (2015) Cystic fibrosis in a retro-positive child. Indian J Pathol Microbiol 58: 204-210.
- Spencer DA, Venkataraman M, Higgins S, Stevenson K, Weller PH (1994) Cystic fibrosis in children from ethnic minorities in the West Midlands. Respir Med 88: 671-675.
- Powers CA, Potter EM, Wessel HU, Lloyd-Still JD (1996) Cystic fibrosis in Asian Indians. Arch Pediatr Adolesc Med 150: 554-555.
- Kapoor V, Shastri SS, Kabra M, Kabra SK, Ramachandran V, et al. (2006) Carrier frequency of F508del mutation of cystic fibrosis in Indian population. J Cyst Fibros 5: 43-46.
- Shah U, Frossard P, Moatter T (2009) Cystic fibrosis: defining a disease under-diagnosed in Pakistan. Trop Med Int Health 14: 542-545.
- Wikipedia contributors (2015) Demographics of India. Wikipedia, The Free Encyclopedia, San Fransico, USA.
- Frizzell RA (1995) Functions of the cystic fibrosis transmembrane conductance regulator protein. Am J Respir Crit Care Med 151: 54-58.
- CFMDB Statistics. Cystic Fibrosis Mutation database.
- Wilmott RW (1998) Making the diagnosis of cystic fibrosis. J Pediatr 132: 563-565.
- Ashavaid TF, Raghavan R, Dhairyawan P, Bhawalkar S (2012) Cystic fibrosis in India: a systematic review. J Assoc Physicians India 60: 39-41.
- Kabra SK, Kabra M, Lodha R, Shastri S (2007) Cystic fibrosis in India. Pediatr Pulmonol 42: 1087-1094.
- Shastri SS, Kabra M, Kabra SK, Pandey RM, Menon PS (2008) Characterisation of mutations and genotype-phenotype correlation in cystic fibrosis: experience from India. J Cyst Fibros 7: 110-115.
- Sachdeva K, Saxena R, Puri R, Bijarnia S, Kohli S, et al. (2012) Mutation analysis of the CFTR gene in 225 children: identification of five novel severe and seven reported severe mutations. Genet Test Mol Biomarkers 16: 798-801.
- Yadav K, Singh M, Angurana SK, Attri SV, Sharma G, et al. (2014) Evaluation of micronutrient profile of North Indian children with cystic fibrosis: a case-control study. Pediatr Res 75: 762-766.
- Agarwal G, Kapil A, Kabra SK, Das BK, Dwivedi S (2005) Characterization of Pseudomonas aeruginosa isolated from chronically infected children with cystic fibrosis in India. BMC Microbiol 21: 5-43.
- Chakrabarty B, Kabra SK, Gulati S, Toteja GS, Lodha R, et al. (2013) Peripheral neuropathy in cystic fibrosis: a prevalence study. J Cyst Fibros 12: 754-760.
- Sharma VK, Raj D, Xess I, Lodha R, Kabra SK (2014) Prevalence and risk factors for allergic bronchopulmonary aspergillosis in Indian children with cystic fibrosis. Indian Pediatr 51: 295-297.
- Moran A, Dunitz J, Nathan B, Saeed A, Holme B, et al. (2009) Cystic fibrosis-related diabetes: current trends in prevalence, incidence, and mortality. Diabetes Care 32: 1626-1631.
- Midha S, Khajuria R, Shastri S, Kabra M, Garg PK (2010) Idiopathic chronic pancreatitis in India: phenotypic characterisation and strong genetic susceptibility due to SPINK1 and CFTR gene mutations. Gut 59: 800-807.
- Sharma H, Mavuduru RS, Singh SK, Prasad R (2014) Increased frequency of CFTR gene mutations identified in Indian infertile men with non-CBAVD obstructive azoospermia and spermatogenic failure. Gene 548: 43-47.
- Farrell PM, Rosenstein BJ, White TB, Accurso FJ, Castellani C, et al. (2008) Guidelines for diagnosis of cystic fibrosis in newborns through older adults: Cystic Fibrosis Foundation consensus report. J pediatr 153: 2-14.
- Lebecque P, Leal T, De Boeck C, Jaspers M, Cuppens H, et al. (2002) Mutations of the cystic fibrosis gene and intermediate sweat chloride levels in children. Am J Respir Crit Care Med 165: 757-761.
- Rosenstein BJ, Cutting GR (1998) The diagnosis of cystic fibrosis: a consensus statement. Cystic Fibrosis Foundation Consensus Panel. J Pediatr 132: 589-595.
- LeGrys VA (2001) Assessment of sweat-testing practices for the diagnosis of cystic fibrosis. Arch Pathol Lab Med 125: 1420-1424.
- LeGrys VA (1996) Sweat testing for the diagnosis of cystic fibrosis: practical considerations. J Pediatr 129: 892-897.
- Menon P (2012) Cystic fibrosis, are we missing in India? Int J Pharm Sci Rev Res 3: 477-481.
- Kabra SK, Kabra M, Gera S, Lodha R, Sridevi KN, et al. (2002) An indigenously developed method for sweat collection and estimation of chloride for diagnosis of cystic fibrosis. Indian Pediatr 39: 1039-1043.
- Schales O, Schales SS (1941) A simple and accurate method for determination of chloride in biological fluids. J Biol Chem 140: 879-884.
- Stern RC, Boat TF, Doershuk CF (1982) Obstructive azoospermia as a diagnostic criterion for the cystic fibrosis syndrome. Lancet 1: 1401-1404.
- Littlewood JM, Wolfe SP (2000) Control of malabsorption in cystic fibrosis. Paediatr Drugs 2: 205-222.
- Daftary A, Acton J, Heubi J, Amin R (2006) Fecal elastase-1: utility in pancreatic function in cystic fibrosis. J Cyst Fibros 5: 71-76.
- Sinaasappel M, Stern M, Littlewood J, Wolfe S, Steinkamp G, et al. (2002) Nutrition in patients with cystic fibrosis: a European Consensus. J Cyst Fibros 1: 51-75.
- Wark P, McDonald VM (2009) Nebulised hypertonic saline for cystic fibrosis. Cochrane Database Syst Rev 2: 001506.
- Derichs N (2013) Targeting a genetic defect: cystic fibrosis transmembrane conductance regulator modulators in cystic fibrosis. Eur Respir Rev 22: 58-65.
- Quittner A, Suthoff E, Rendas-Baum R, Bayliss MS, Sermet-Gaudelus I, et al. (2015) Effect of ivacaftor treatment in patients with cystic fibrosis and the G551D-CFTR mutation: patient-reported outcomes in the STRIVE randomized, controlled trial. Health Qual Life Outcomes 13: 93-101.
- Kuk K, Taylor-Cousar JL (2015) Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: current evidence and future prospects. Ther Adv Respir Dis 9: 313-326.
- Wilschanski M, Miller LL, Shoseyov D, Blau H, Rivlin J, et al. (2011) Chronic ataluren (PTC124) treatment of nonsense mutation cystic fibrosis. Eur Respir J 38: 59-69.
- Patrick Lebecque (2012) The Prognosis of Cystic Fibrosis - A Clinician’s Perspective. In: Sriramulu D (ed.). Cystic Fibrosis-Renewed Hopes Through Research. Intech, Rijeka, Croatia.
- Gild R, Clay CD, Morey S (2010) Aquagenic wrinkling of the palms in cystic fibrosis and the cystic fibrosis carrier state: a case-control study. Br J Dermatol 163: 1082-1084.
Citation: Mandal A, Kabra SK, Lodha R (2015) Cystic Fibrosis in India: Past, Present and Future. J Pulm Med Respir Res 1: 002.
Copyright: © 2015 Anirban Mandal, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.