E. coli BL21 Bacterial and Newcastle Pestikal lasota Viral Ghosts Preparation, DNA/RNA Elimination/Purification/Isolation/ Quantification Using Agarose Gel Entrapment Bubble Method
*Corresponding Author(s):
Amro Abd Al Fattah AmaraDepartment Of Protein Research, Genetic Engineering Biotechnology Research Institute. City Of Scientific Research And Technological Applications, Alexandria; Universities And Research Center District, New Borg El-Arab , Egypt
Tel:+20 34593422,
Email:amroamara@web.de
Abstract
Sponge like protocol is a new protocol able to turn most microbes including viruses to empty cells or empty envelops. The original protocol guarantees full evacuation, however, in some cases it need a validation. In this study, simple tools are described to separate the genetic constituents of both partially prepared E. coli BL21 and Newcastle Pestikal lasota strain ghosts. The used methods describe the preparation of entrapment bubble (in the agarose gel) as a proposed step for separating the genetic materials from the other constituents. That might solve some technical problems particularly during the virus ghost preparation. The removed DNA and/or RNA by the agarose have been collected. Different simple methods have been used in subsequent steps such as the electrophoresis using the electrical current alone or with the dialysis bag, and the double Eppendorf tube centrifugation method. The DNA and/or RNA have been measured spectrophotometrically using quartzes cuvette at 260nm. This study is a new tool for separating the genetic elements from the microbial ghosts. The method could be either used for the evacuation processes, DNA RNA isolation and purification and for concentrating virus (es)/ ghost virus (es). It could be used also in other technical applications such industrial and pharmaco/medical applications etc.
Keywords
Agarose gel; E. coli BL21 ghost cells; Microbial ghosts; Newcastle Pestikal lasota ghost
ABBREVIATIONS
MGC: Minimum Growth Concentration
MIC: Minimum Inhibition Concentration
RBC: Red Blood Cells
SDS: Sodium Dodecyl Sulfate
SL: Sponge Like
SLP: Sponge Like Protocol
INTRODUCTION
The ancient Egyptian might be the first how succeeded to completely evacuating cells from their soluble contents, a process known as mummification. It might be that the first remarked known ghost cell (s) is existed in the first sample examined under the first microscope by Antonie van Leeuwenhoek [1]. That because microbial ghosting is a natural phenomenon [1]. In fact, we turning microbes to ghosts or lysis them daily by attacking the microbes which invade our bodies or inter to it normally, during the feeding process or by other routes. An example is the lysozyme in our mouth’s saliva which is a part of our innate immune system.
Weibull (1956) is succeeded to prepare Bacillus megaterium as protoplast. Its protoplast has been given the name “ghosts” [2]. The phage ghosts and bacterial ghosts have been known in the middle of the 19th century because of the development of the molecular biology and the microscopes [3]. Red Blood Cells (RBCs) ghosts have been also prepared. Other forms of different cells have the ghosts feature have been described [4]. The concept of bacterial lysis by the bacteriophage E lysis protein (from a cloned bacteriophage ΦX174 gene) leads to establish a protocol restricted to the gram negative ghost cells [5-8]. To avoid pathogenic islands and antibiotic resistance genes in the BGs vaccine preparation, a nuclease degraded the DNA [9,10].
A new protocol proves success with all types of microbes has been introduced. It has been given the name Sponge Like (SL) protocol, which is derived from using the critical chemical concentration of some active chemical compounds or the critical activity of some proteins and enzymes. Some physical and biological parameters that could induce evacuation has been used [11]. SL protocol found acceptance and proved to be efficient when applied to different microbes [11-17]. The prepared bacterial cells using the SL protocol are maintained correct cell structures and surface antigens.
Lysozyme is an enzyme able to lysis most of the bacterial strains and it has been used in the original SL protocol [11]. Harsh extermothermophilic spore former, protease and keratinase producer Bacillus stearothermophilus strain has been prepared as ghost cells using the minimum activity concentration of the native/purified hen white egg lysozyme [11,18,19]. Using the bio-critical concentration of a single compound such as H2O2 is enables the evacuation of the Newcastle virus from their RNA constituents using the same H2O2 concentration which applied to E. coli JM109 and E. coli BL21 [20]. The bio-critical concentration of H2O2 with the E. coli strains has been in the range and succeeded to cure the Newcastle virus from its RNA [20]. It is of great important to keep the body oxidant in certain limit, and not deteriorate them totally by over use of the antioxidant [21].
Recently a fast plasmid slot lysis protocol has been described by Amara (2018) for DNA and plasmid isolation and ghost cells preparation [22]. Some other excellent protocols describe different type of DNA preparation [23-26]. The agarose gel electrophoresis causes a migration for the plasmids as well as their DNA contents. That enables the determination of the cells which have the right clones. In fact, the protocol has been used as an alternative solution for the white/blue selection [22,27]. Only the lysis buffer is applied for a short time. The protocol is used for microbial ghosts’ preparation using the electrophoresis current to force the cells evacuation and the process, substitute the centrifugation and the washing steps in the original protocol [22,27].
Different exopolysaccharides have been used in various molecular and biotechnological applications. Agarose is a linear natural polymer with a molecular weight of about 120,000, consisting of alternating D-galactose and 3,6-anhydro-L-galactopyranose [28,29]. It is usually used for DNA and RNA isolation and sometimes used instead of the agar to measure microorganism motility and mobility.
This study aims to entrap the partially prepared bacterial/virus ghosts in a prepared agarose gel bubbles, and to eliminate/ purify/ isolate/ quantify their DNA and/or RNA contents.
MATERIALS AND METHODS
Microbial strains
Bacterial strain
coli BL21 recombinant cells (Promega) has been used in this study. The E. coli contains a plasmid pLysS. The E. coli genotype is pLysS strain F’, ompT, gal [dcm], [lon], hsdSB, (rB–mB-), gal, dcm, rne131, (DE3), pLysS (Camr) [30,31].
Viral strain
Newcastle Pastikal lasota SPF virus (Hyper V-for animal health -GENER) strain obtained from local pharmacy has been used in this study. It has been preserved at -20°C at a refrigerator.
Preparation of bacterial ghost cells
coli BL21 prepared as ghost cells following Amara et al (2013) [11,15,16]. Only NaHCO3 has been used instead of CaCO3 as described first by Amara (2015) [17].
Preparation of viral ghost
Newcastle Pastikal lasota virus has been prepared following the methods described by El-Baky and Amara (2014) [20].
PREPARATION OF TRAPPED AGAROSE BUBBLE
The trapped bubble formation
1.0% (w/v) agarose gels solution have been used where one gram of the agarose (Sigma-Aldrich, Steinheim, Germany) has been dissolved by boiling it in 100 ml TBE buffer (50 mM Tris/HCl, 50 mM Boric acid, 2.5 mM EDTA, pH 8.5) or in distilled water. The mixture has been heated till complete dissolving of the agarose and left in room temperature to reduce its temperature.
For preparing trapped bubbles, the dissolved agarose has been loaded in a plastic spectrophotometer cuvette and the agarose left till becoming viscous. Sterile syringe with a fine needle has been used. The syringe which contains air has been left 5 min in -20°C to cool the air inside. The syringe needle then has been interred in the agarose (it is recommended to use fine needle), then the cold air has been injected gently to form a single bubble. The syringe needle then removed gently to avoid the loosing of the formed bubble. The agarose left till complete dryness.
Removing of the agarose from the spectrophotometer cuvette
The down side of the cuvette is removed (using a cutter). The agarose is removed from the cuvette by pressing it gently using suitable tool. Alternatively, symmetric cuvette could be used and the bottom side only could be removed.
Sample loading in the trapped bubbles
The prepared sample, each has been injected to a single bubble till complete removal of the inside-air. The injected volume has been calculated. The needle is removed. In case of injecting the sample before removing the cuvette body, after the injection step the agarose is turned 90 degrees to keep the injected surface up. Loading dye could be added just to prove the immigration of the DNA and/or RNA, but in experiments aim to isolate native DNA and/or RNA, it is recommended to calculate the time of the DNA and/or RNA immigration or using the additional step (the dialysis page or the alternative step the double Eppendorf tubes centrifugation method) as below. The agarose containing the samples are put in a DNA electrophoresis unit (MAX FIL TUG-SYS unit Cambridge-Science Park) and covered gently with the running buffer. The gel electrophoresis electrical current then has been run at 70 V for 2-3 min (or as calculated). Both of the agarose concentration or the current electrical power could be increased or decreased to appropriate level based on the used samples. For experiments aiming to investigate only the presence of the DNA and/or RNA or plasmid, 70% glycerol can be improving the samples loading.
The DNA and/or RNA collection
After the migration of the DNA/RNA in the gel there are two simple methods have been used to collect the DNA and/or RNA. The first is the dialysis page and the second is the double Eppendorf tubes centrifugation method for collecting the DNA and/or RNA.
Isolation of DNA and/or fragments using dialysis bag
Dialysis page (SERVA-VISKINGR) with a diameter equal to 16 nm was prepared according to the manual included. The agarose gel containing the sample and the migrated DNA and/or RNA are transferred to a treated dialysis bag contains the running buffer. The bag then closed well and put in electrophoretic chamber and the electric current is allowed (70 V). The estimated running time is calculated regarding to the size of the gel. The DNA will pass through the gel to the buffer due to the effect of electric current. DNA can be attached to the surface of the dialysis bag, however by putting the bag in the opposite direction and allowing the electrophoretic current 70 V for 5 sec., that should be enough to detach the DNA and/or RNA from the surface of the dialysis bag. The DNA and/or RNA then collected by opening the bag and collecting the buffer.
Isolation of DNA and/or RNA using duple Eppendorf tubes centrifugation method
Under aseptic conditions, two Eppendorf tube are used one with small pore in its bottom covered by sterile filter paper. The migrated DNA and/or RNA agarose gel part could be cut to small slices (after full DNA and/or RNA migration) or the whole used gel which contains the sample can be used. The gel slice is putted inside the Eppendorf which contains a poor and the filter paper, then both are put in a second normal Eppendorf. The two Eppendorf are centrifuged at 4000 rpm/10 min then the speed has been elevated to 13000 rpm for 5 min (Biofuge 13, Heraeus instrument Christ, Osterode). The DNA and/or RNA will be separated from the agarose gel and will be passed through the filter paper and then through the Eppendorf poor and collected in the second Eppendorf. The centrifugation forces and time could be reduced or increased to appropriate level based on the sample type.
Visualization of the DNA and/or RNA on the gel
After electrophoresis, the gel has been stained in ethidium bromide solution for at least 10 min, followed by detecting the DNA and/or RNA under UV light at 260 nm in a UV transilluminator (SYNGENE-BIO IMAGEREADY). An image has been generated.
Determination of the DNA Concentration
The concentration of the DNA has been determined spectrophotometrically (Libra S70-Biochrom Cambridge) by measuring the absorption at 260 nm. Quartz cuvette has been used. The absorbance has been set to 0 using the running buffer as a blank ample. An extinction E260 = 1 corresponds to 50 µg dsDNA ml-1 [32]. And 38.5-40 µg ss DNA/RNA ml-1.
Additionally, the concentration of DNA has been estimated by following the fluorescence of bands within the agarose gel that had been re-stained with ethidium bromide. The amount of DNA which has been just visible after electrophoresis, staining and photography on the gel has been set to 2 ng.
Collection of the Newcastle Pastikal lasota virus and the E. coli BL21 from the agarose gels.
After separating the DNA and/or RNA the ghosts of the Newcastle Pastikal lasota virus and the E. coli BL21 were collected by opening the agarose passing inside the bubbles and its content has been separated for each. Gentle vortex at 40 rpm for the agarose sample before the cut could improve the process. The collection can be done in different point of the protocol based on the nature of the preparation.
RESULTS AND DISCUSSION
Isolating the genetic elements from the different microbes is a basic tool in the molecular biology research. Understanding the behaviors of the different genetic elements will facilitate their isolation safely and correctly. Simplifying such tools is in demand particularly in the developmental countries where the cost of an experiment might be a critical issue [27,33-36]. Many simple enzymatic and non-enzymatic based methods have been described for either the isolation of the DNA, RNA or the plasmid DNA [27,33-36].
Bacterial ghosts are intact, inactive, evacuated bacterial cell with correct 3D structure and surface antigens. Ghost techniques increase the safety of the killed vaccines, while maintaining their antigenicity due to mild preparation procedures. SL protocol is, guarantee an inactive microbial cells with correct surface antigen and 3D structure. Ghost cells may express and/or carry several expressed, surface antigens, plasmid DNA carrier, drug as delivery system, adjuvants, and dendritic cell inducer. Unlike attenuation, inactivation of microbes is based on complete killing for the microbes using chemical, biological or physical tools. However, the SL protocol is a fast, cheap and in house protocol for microbial cell evacuation [11,15-17]. It describes bacterial cell evacuation from its cytoplasmic contents. It has been optimized using Plackett-Burman design to map the best cells’ evacuation experimental conditions, which give the best un-deformed 3D structure and un-deteriorated surface antigens [11,15]. Then the best experimental conditions are used to fasting the protocol time [11]. Its main concept is to determine the Minimum Inhibition Concentration (MIC) and the Minimum Growth Concentration (MGC) of chemical compounds able to induce pores in the cell walls [1,11-17,19,37-39]. The used chemical compounds until now are Sodium Dodecyl Sulfate (SDS), NaOH, NaHCO3, H2O2, CaCO3, NaCl and ethanol. Each of those compounds has been selected based on their effect on the cell wall, genetic elements or the protein contents [28,40-52]. In addition, 60% ethanol has been used to improve the DNA extraction processes [11]. Apparently, the original protocol published by Amara et al. (2013), has been the first protocol describes the use of the MIC and the MGC of the used chemical compounds for ghost cells preparations. In the original protocol only one colony is survived and the whole experiment batch has been degraded by activating the lysozyme gene using chloramphenicol [11]. However, the main prepared ghost cells have been prepared from the highest quality yield experiments as reintroduced in the reduced protocol [11]. Controlling the cells 3D structure during their evacuation steps using the light microscope guarantee full evacuation with nearly no effect on the 3D or the surface antigens. For each microbe there is a need for doing some adjustments, that due to the differences in the cell wall or envelops/cote constituents, but after adjusting the evacuation conditions, the protocol is reliable and straightforward.
The idea of the protocol has been used to evacuate the Newcastle virus [20]. Recently, the virus has been prepared as empty viral ghost using the bio-critical concentration of the H2O2 [21]. Newcastle virus (LaSota strain-RNA virus) has been used as a model [21]. It is a RNA virus. The images of the agarose gel as well as the spectrophotometer spectra at 260/280nm have been proved successful release and degradation to the virus RNA [21].
Other microbes such as the eukaryotic Saccharomyces cerevisiae, Aspargillus nige, Aspargillus flavus have been prepared as ghosts. [53-55], additionally the oyster mushroom’s spores have been turned as ghosts [56]. The first drug delivery system using the SLP with the gossypol acetic acid and E. coli and Saccharomyces cerevisiae has been published by Amara (2015) [53].
To prove that the protocol concept is universal and could exceed the chemical compounds to the biological ones, lysozyme and proteinase K have been used with the spore former extremothermophilic B. stearothermophilius [18]. The enzymes when used in their minimum killing activity concentrations and their minimum living activity concentrations succeeded to turn the spore former bacteria to ghost cells [18]. It enables the control of the pore formation by many controlling factors, variables and steps. One could say that this protocol and its concept enable the evacuation of the biological cells or viruses without harming their cell wall/virus cote, or their outer surface antigen [37-39].
Many technical solutions have been discussed in the different published papers where each microbe considers as case by case study. The SL protocol not deals only with the cell deactivation but also deal with its complete evacuations. Despite using the electron microscope, the protocol considers to be in a house tool for microbial ghost formation. One technical problem has been deviated the protocol to use some sophisticated instruments, It is the virus collection with constant amount constant during the ghost preparation steps, particularly during the separation of their genetic elements from their cote.
This study has been modified to be able to isolate the DNA, RNA and other cell components, as well as the microelements that could pass from the agarose gel during the electrophoresis without changing the cell load concentration. Additionally, it could be used for different target experiments include ghost cells preparation, DNA/RNA/protein isolation etc.
Different methods have been identified the isolation of the DNA and the RNA from the different microbial strains. However, in most cases such as the alkaline lysis protocol for the plasmid preparation or the phenol protocol for the DNA preparation the protocols usually did not care with the other components of the microbial cells. Recently, Amara describe a protocol for fast slot lysis for DNA and plasmid in agarose gel separation with the recommendation to calculate the used amount in the lysis buffer to produce bacterial ghost instead of lysis the cells [22,27]. In addition, the study highlighted the use of dialysis page to separate the DNA from the bacterial cell either directly or after being loaded in the agarose.
However, in fine microbes like viruses the loading of the virus in the normal slab agarose gel electrophoresis did not granite the retrieve of the virus from the agarose. It is of great important and a technical demand to keep the virus load constant during the molecular biology protocols and experiments. One major technical limiting factor is the virus load. This study succeeded to retrieve both of the bacterial and viral ghosts without changing their load because they are entrapped in the agarose bubbles (data not shown). More studies are in need to prove that using quantitative experiments.
Even Amara highlighted the important of using DNases and RNases in the ghost cell preparation but those enzymes will need additional steps for their removing. The methods have been described in this study did not describe any use of additional proteins or enzymes, it is an additional step toward better bacterial and viral ghost coats preparation, DNA/RNA/Plasmid isolations, and the various described techniques. The data obtained from the different concentration of the isolated DNA and/or RNA of E. coli BL21 ghosts (DNA and/or RNA) and Newcastle Pestikal lasota viral ghosts (RNA) using the Dialysis pages and the Double centrifugation methods for DNA and/or RNA isolation as in (Table 1) show various DNA and/or RNA isolated quantities. By analysis the data it is showing that the major quantities of the DNA and/or RNA are isolated during the Dialysis page step (Table 1) and (Figure 1) The quantity of the isolated RNA from the virus is less than the DNA and/or RNA isolated from the E. coli BL21(Table 1) and (Figure 1) (step 7 and 8). That could be either explained that the used amount of the originally used virus load contains RNA less than that used for bacterial load. Or, the virus is very sensitive to the treatment with H2O2. Additionally, it might be that the first migration step need more adjustment while the stained agarose gels show less viral RNA migrated to the bottom of the gel (Figure 1) (step 6). It is a fact that the RNA is migrated faster than the DNA. Agarose in % more than 1% will give better results. The experimental conditions did not enable the migration of the protein might have existed in the sample through the agarose, however more validation studies are needed in the future work. Meanwhile, increasing the agarose concentration will fervor better pure DNA/RNA. In contrast, using this protocol in a lower agarose concentration enable protein purification [Data not shown] [57]. Or the DNA and the RNA samples should be running separately. However, the protocol shows efficient removal for the DNA and/or RNA. This study recommended the following points:
- • Specifying the target of the protocol, either being for the DNA and/or RNA isolation/purification/ quantification, ghosts validation, ghosts preparation etc. and based on the target the protocol must be directed and some of its steps could be removed or changed.
- • Running each of the bacterial ghosts and the virus ghosts separately.
- • Using agarose higher than 1% in case of the RNA samples.
- • Eliminating the first DNA and/or RNA migration step for better DNA and/or RNA isolation.
- • Using standard DNA as a control.
- • Using equal side’s plastic cuvettes for the entrapped bubbles preparation.
- • Using voltage equal to 70V or less. For better sample collection lesser volt should be used.
- • Detaching the DNA and/or RNA from the dialysis page after their separation using the electrical current.
- • The method could be used for concentrating the ghosts and reduce their suspended volume by repeating the process or by using a bigger entrapped bubble, or pre-prepared unit.
|
Sample name |
Calculated amount of DNA/RNA |
|||
|
µg ml-1 |
Total µg ml-1
|
|||
|
Dialysis page |
Double centrifugation |
|||
|
Running buffer |
Set to 0 |
Set to 0 |
0 |
|
|
DNA marker |
365.5 |
151.04 |
|
|
|
E. coli BL21 ghosts DNA/RNA |
328.7 |
143.84 |
472.54 |
|
|
Newcastle Pestikal lasota viral ghosts RNA |
123.36 |
63.2 |
|
|
Table 1: Different concentration of the isolated DNA/RNA of E. coli BL21 ghosts (DNA/RNA) and Newcastle Pestikal lasota viral ghosts (RNA) using the Dialysis pages and the Double centrifugation methods for DNA/RNA isolation.
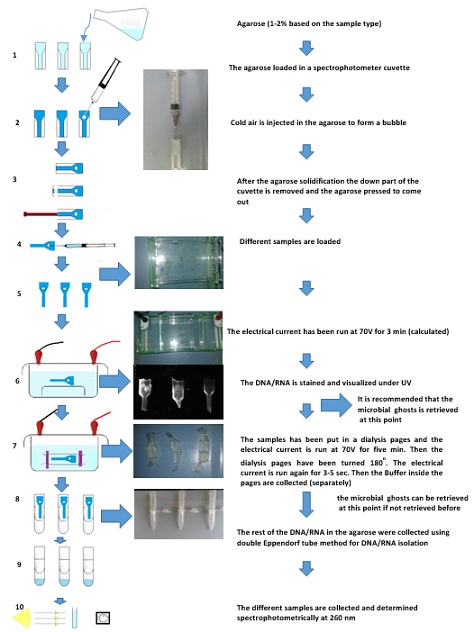 Figure 1: Diagram showing different steps in E. coli BL21 bacterial and Newcastle Pestikal lasota viral ghosts preparation, DNA/RNA elimination/ purification/ isolation/ quantification using agarose gel entrapment bubble method: 1) Agarose preparation; 2) Entrapped bubbles formation in the agarose gel inside the plastic cuvette as container; 3) removing of the agarose from the cuvette; 4) Sample injection in the entrapped bubbles; 5) adjusting the direction of the agarose contains loaded sample(s) 6) Electrophoresis for the samples, for migrating the DNA/RNA; 7) dialysis using electrophoresis current (for the samples) for collecting the DNA/RNA; 8) double Eppendorf tube centrifugation method for collecting the DNA/RNA; 9) The collected DNA/RNA; 10) Spectrophotometer determination for the samples at 260nm - The stained agarose gels show: a) DNA sample marker; b) E. coli BL21 bacterial DNA/RNA; c) Newcastle Pestikal lasota viral RNA - The E. coli BL21 bacterial and Newcastle Pestikal lasota viral ghosts can be collected in two points (the first one is recommended).
Figure 1: Diagram showing different steps in E. coli BL21 bacterial and Newcastle Pestikal lasota viral ghosts preparation, DNA/RNA elimination/ purification/ isolation/ quantification using agarose gel entrapment bubble method: 1) Agarose preparation; 2) Entrapped bubbles formation in the agarose gel inside the plastic cuvette as container; 3) removing of the agarose from the cuvette; 4) Sample injection in the entrapped bubbles; 5) adjusting the direction of the agarose contains loaded sample(s) 6) Electrophoresis for the samples, for migrating the DNA/RNA; 7) dialysis using electrophoresis current (for the samples) for collecting the DNA/RNA; 8) double Eppendorf tube centrifugation method for collecting the DNA/RNA; 9) The collected DNA/RNA; 10) Spectrophotometer determination for the samples at 260nm - The stained agarose gels show: a) DNA sample marker; b) E. coli BL21 bacterial DNA/RNA; c) Newcastle Pestikal lasota viral RNA - The E. coli BL21 bacterial and Newcastle Pestikal lasota viral ghosts can be collected in two points (the first one is recommended).
CONCLUSION
The SL protocol guarantees full microbial evacuation. This study is an additional step for the original protocol evaluation, DNA.RNA separation, examining the ghost cells quality, concentrating the ghost cells and validates ghost cells free DNA and/or RNA. Successfully, this preliminary study introduces for the first time, a method where the microbes did not being lost or their load is diluted during the agarose electrophoresis with subsequent safe steps for isolation of their DNA and/or RNA contents.
CONFLICT OF INTEREST
The author declares that there is no conflict of interest of any kind.
FUNDING
Institutional support.
ACKNOWLEDGEMENT
The author acknowledges the DAAD for the PhD. scholarship provided to the author during 1999 to 2003 which support his knowledge in the molecular biology and lead to finalize this study.
REFERENCES
- Amara A.A (2015) Kostenlos Viral Ghosts, Bacterial Ghosts, Microbial Ghosts and more, Schüling Verlage
- Weibull, C (1956) the nature of the ghosts obtained by lysozyme lysis of Bacillus megaterium, Exp Cell Res 10: 214-221.
- Duckworth DH, Bessman MJ (1965) Assay for the Killing Properties of T2 Bacteriophage and Their “Ghosts”, J Bacteriol 90: 724-728.
- Ansorge I, Benting J, Bhakdi S, Lingelbac K (1996) Protein sorting in Plasmodium falciparum-infected red blood cells permeabilized with the pore-forming protein streptolysin O. Biochem J 315: 307- 314.
- Dong H, Han X, Bai H, He L, Liu L, et al. (2012) Mutation of lambdapL/pR-cI857 system for production of bacterial ghost in Escherichia coli. Sheng Wu Gong Cheng Xue Bao 28: 1423-1430.
- Eko FO, Witte A, Huter V, Kuen B, Furst-Ladani S, et al. (1999) New strategies for combination vaccines based on the extended recombinant bacterial ghost system. Vaccine 17: 1643-1649.
- Hu J, Zuo J, Chen Z, Fu L, Lv X, et al. (2019) Use of a modified bacterial ghost lysis system for the construction of an inactivated avian pathogenic Escherichia coli vaccine candidate. Vet Microbiol 229: 48-58.
- Lubitz W, Witte A, Eko FO, Kamal M, Jechlinger W, et al. (1999) Extended recombinant bacterial ghost system. J Biotechnol 73: 261-273.
- Hutchison III CA, Sinsheimer RL (1966) The process of infection with bacteriophage phi-X174. X. Mutations in a phi-X Lysis gene. Journal of Molecular Biology 18: 429-447.
- Haidinger W, Mayr UB, Szostak MP, Resch S, Lubitz W (2003) Escherichia coli ghost production by expression of lysis gene E and staphylococcal nuclease. Applied and Environmental Microbiology 69: 6106-6113.
- Amara AA, Salem-Bekhit MM, Alanazi FK (2013) Sponge-like: A new protocol for preparing bacterial ghosts. The Scientific World Journal 2013: 545741.
- Amara AA, Neama AJ, Hussein A, Hashish EA, Sheweita SA (2014) Evaluation the surface antigen of the Salmonella typhimurium ATCC 14028 ghosts prepared by “SLRP”. The Scientific World Journal 2014: 840863
- Menisy M, Hussein A, Ghazy AA, Sheweita S, Amara AA (2017) Klebsiella pneumoniae Ghosts as vaccine using sponge like reduced protocol. Cellular Mole. Med 3: 1-8.
- Sheweita SA, Batah AM, Ghazy AA, Hussein A, Amara AA (2019) A new strain of Acinetobacter baumannii and characterization of its ghost as a candidate vaccine. Journal of Infection and Public Health 12: 831-842.
- Amara AA, Salem-Bekhit MM, Alanazi FK (2013) Plackett-Burman randomization method for Bacterial Ghosts preparation form E. coli JM109. Saudi Pharmaceutical Journal 22: 273-279.
- Amara AA, Salem-Bekhit MM, Alanazi FK (2013) Preparation of bacterial ghosts for E. coli JM109 using sponge-like reduced protocol. Asian J Biol Sci 6: 363-369.
- Amara AA (2015) Saccharomyces cerevisiae Ghosts using the sponge-like re-reduced protocol. SOJ Biochem 1: 1-4.
- Amara AA (2016) The critical activity for the cell wall degrading enzymes: Could the use of the lysozyme for microbial ghosts preparation establish emergence oral vaccination protocol. International Science and Invastigation Journal 5: 351-369.
- Amara AA (2016) Lysozymes, proteinase K, bacteriophage E lysis proteins, and some chemical compounds for microbial ghosts preparation: a review and food for thought. SOJ Biochem 2: 1-16.
- Abd El-Baky N, Amara AA (2014) Newcastle disease virus (LaSota strain) as a model for virus Ghosts preparation using H2O2 bio-critical concentration. International Science and Investigation journal 3: 38-50.
- Amara AA (2010) The philosophy behind exo/endo/existing antioxidants and our built-in oxidant and antioxidant system. Pharmazie 65: 711-719.
- Amara AA (2018) Fast Plasmid Slot Lysis and Gram-Negative Bacteria Ghost Preparation Protocol. Austin J Proteomics Bioinform & Genomics 5: 1-1025.
- Green MR, Sambrook J (2018) Preparation of Plasmid DNA by Alkaline Lysis with Sodium Dodecyl Sulfate: Maxipreps, Cold Spring Harb Protoc 2018.
- Sambrook J, Russell DW (2006) Preparation of Plasmid DNA by Alkaline Lysis with SDS: Maxipreparation. CSH Protoc 1: 1-2006.
- Sambrook J, Russell DW (2006) Removal of Small Fragments of Nucleic Acid from Preparations of Plasmid DNA by Centrifugation through NaCl. CSH Protoc 1:1- 2006.
- Sambrook J, Russell DW (2006) Extraction of Bacteriophage lambda DNA from Large-scale Cultures Using Proteinase K and SDS. CSH Protoc 1: 1-2006.
- Amara AA (2005) Simple slot lysis and plasmid curing for determination the transfer of resistant factors within the pathogenic microbial strains. In Proceeding of Tagungsband zum 2. Gemeinsamen kongress der DGHM und VAAM Göttingen, Germany, BioSpectrum, 74.
- Amara AA, Hassan MA, Abulhamd AT, Haroun BM (2011) algs genes: H2O2 Map their Viability and Validity in Pseudomonas aeruginosa. IJBB 7: 321-330.
- Tombs M, Harding SE (1998) An introduction to polysaccharide biotechnology. Taylor and Francis 141-144.
- Studier FW, Moffatt BA (1986) Use of bacteriophage T7 RNA polymerase to direct selective high-level expression of cloned genes. Journal of Molecular Biology 189: 113-130.
- Davanloo P, Rosenberg AH, Dunn JJ, Studier FW (1984) Cloning and expression of the gene for bacteriophage T7 RNA polymerase. Proceedings of the National Academy of Sciences of the United States of America 81: 2035-2039.
- Sambrook J, Fritsch EF, Mainiatis T (1989) Molecular Cloning a Laboratory Manual. Cold Spring Harbor Laboratory, (2nd edition) Cold Spring Harbor, NY, USA,
- Amara AA, Afifi IK, Younis MA, Sharaf MM, Shabeb MS (2005) Non-Enzymatic methods for DNA isolation from different microbial strains. J. Biotechnol 21: 339-349.
- Amara AA, Afifi IK, Younis MA, Sharaf MM, Shabeb MS (2005) Simple methods for microbial DNA isolation. In Proceeding of Tagungsband zum 2. Gemeinsamen kongress der DGHM und VAAM Göttingen, Germany, BioSpectrum 35.
- Amara AA (2007) Introduction to the basic molecular microbiology protocols- (First edition). Schüling Verlage, Germany.
- Amara AA (2010) Introduction to the basic molecular microbiology protocols- (Second edition). Schüling Verlage Germany.
- Amara AA (2017) Cracking the Microbial Cell Wall. SOJ Vaccine Research 6: 363-369.
- Amara AAAF (2017) An invitation: Case-by-Case Cell Ghosts preparation, International Science and Investigation journal 6: 1-7.
- Amara AA (2017) Smart green technology for microbial ghosts preparation, IIOAB JOURNAL 8: 53-54.
- Armeni T, Battino M, Stronati A, Pugnaloni A, Tomassini G, et al. (2001) Total antioxidant capacity and nuclear DNA damage in keratinocytes after exposure to H2O2. Biol Chem 382: 1697-1705.
- Feldberg RS, Carew JA, Paradise R (1985) Probing Cu(II)/H2O2 damage in DNA with a damage-specific DNA binding protein. J Free Radic Biol Med 1: 459-466.
- Iliakis GE, Pantelias GE, Okayasu R, Blakely WF (1992) Induction by H2O2 of DNA and interphase chromosome damage in plateau-phase Chinese hamster ovary cells. Radiat Res 131: 192-203.
- Midorikawa K, Kawanishi S (2001) Superoxide dismutases enhance H2O2-induced DNA damage and alter its site specificity. FEBS Lett 495: 187-190.
- Agarwal RK, Perl A (1993) PCR amplification of highly GC-rich DNA template after denaturation by NaOH. Nucleic Acids Res 21: 5283-5284.
- Broad TE, Forrest JW, Pugh PA (1988) Effect of NaOH on hybridization efficiency of DNA. Trends Genet 4: 1-146.
- Kadokami Y, Deike CA, Lewis RV (1995) Precipitation of the template DNA can be omitted after NaOH denaturation in double-stranded DNA sequencing. Biotechniques 18: 40-41.
- Rigaud G, Grange T, Pictet R (1987) The use of NaOH as transfer solution of DNA onto nylon membrane decreases the hybridization efficiency. Nucleic Acids Res 15: 1-857.
- Schneeberger RG, Gorman SW, Cullis CA (1988) Effect of NaOH on hybridization efficiency of southern-transferred DNA. Trends Genet 4: 1-328.
- Mrsa V, Tanner W (1999) Role of NaOH-extractable cell wall proteins Ccw5p, Ccw6p, Ccw7p and Ccw8p (members of the Pir protein family) in stability of the Saccharomyces cerevisiae cell wall. Yeast 15: 813-820.
- Amara AA, Steinbüche A (2013) New medium for pharmaceutical grade Arthrospira, International Journal of Bacteriology 2013: 1-9.
- Amara AA (2005) Biochemical and Molecular Characterization of PHA synthases from Pseudomonas aeruginosa, Ralstonia eutropha, Aeromonas punctata as well as of the (R)-3-hydroxyacyl-ACP: CoA transacylase from Pseudomonas putida (PhD. degree at 2003) Schüling Verlage Germany.
- Abrao MG, Billerbeck AE, Nishi MY, Marui S, Mendonca BB (2005) Standardization of DNA extraction with NaCl from oral mucosa cells: application in PROP1 gene study. Arq Bras Endocrinol Metabol 49: 978-982.
- Amara AA (2015) Bacterial and Yeast Ghosts: E. coli and Saccharomyces cerevsiae preparation as drug delivery model. International Science and Investigation Journal 4: 11-22.
- El-Baky NA, Sharaf MM, Amer E, Kholef HR, Amara AA, et al. (2018) The minimum inhibition and growth concentration for controlling fungal infection as well as for ghost cells preparation: Aspargillus flavus as a model, Biomedical Journal of Scientific and Technical Research 10: 1-5.
- El-Baky AN, Sharaf MM, Amer E, Kholef HR, Amara AA, et al. (2018) Protein and DNA isolation from Aspergillus niger as well as ghost cells formation. SOJ Biochem 4: 1-7.
- Haddad A, Sharaf MM, Kenawy AMA, Amara AA (2019) Oyster Mushroom Spores Ghost Preparation for Medicinal, Biotechnological and Forensic Applications Biomed J Sci & Tech Res 24: MS.ID.003994.
- Amara AA (2020) Could agarose gel electrophoresis being used for protein purification: A study. Under preparation.
Citation: Amara AA (2020) E. coli BL21 Bacterial and Newcastle Pestikal lasota Viral Ghosts Preparation, DNA/RNA Elimination/Purification/Isolation/Quantification Using Agarose Gel Entrapment Bubble Method. J Protein Res Bioinform 2: 010.
Copyright: © 2020 Amro Abd Al Fattah Amara, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.