Entecavir Patent Evaluation & Genotoxicity
*Corresponding Author(s):
Krishnasarma PathyIpl Research Centre, Lucknow, India
Tel:+91 9919188895,
Email:drkrishnasarmapathy@yahoo.in
Abstract
Entecavir is an oral antiviral drug used in the treatment of hepatitis B infection. Entecavir is a guanosine nucleoside analogue with selective activity against Hepatitis B Virus (HBV), which inhibits reverse transcription, The initial patent on entecavir expired in South Africa in 2011 ZA 1991/07894. Entecavir, a deoxyguanosine analog, is one of the most widely used oral antiviral NAs against hepatitis B virus [1]. It has reported that entecavir gave positive responses in both genotoxicity and carcinogenicity assays. However the genotoxic mechanism of entecavir remains elusive. To evaluate the genotoxic mechanisms, we analyzed the effect of entecavir on a panel of chicken DT40 B-lymphocyte isogenic mutant cell line deficient in DNA repair and damage tolerance pathways. Our results showed that Parp1-/- -/- mutant cells defective in Single-Strand Break (SSB) repair were the most sensitive to entecavir. Brca1-/- , Ubc13-/- and translesion-DNA-synthesis deficient cells including Rad18-/- and Rev3-/- were hypersensitive to entecavir. XPA-/- mutant deficient in nucleotide excision repair was also slightly sensitive to entecavir. γ-H2AX foci forming assay confirmed the existence of DNA damage by entecavir in Parp1-/- -/-, Rad18-/- and Brca1-/- mutants. Karyotype assay further showed entecavir-induced chromosomal aberrations, especially the chromosome gaps in Parp1-/- -/-, Brca1-/- , Rad18-/- and Rev3-/- cells when compared with wild-type cells. These genetic comprehensive studies clearly identified the genotoxic potentials of entecavir and suggested that SSB and postreplication repair pathways may suppress entecavir-induced genotoxicity.
Keywords
INTRODUCTION
Hepatitis B disease is a major health problem in the world, today received FDA approval for the treatment of many nucleoside analog drugs are used. Entecavir in our study is one of them, there is much work related to genotoxicity. To determine the entecavir the genotoxic effects in our study, people in peripheral lymphocyte cells in vitro culture medium entecavir three different concentrations (1.66mg/mL, 3.33mg/mL and 6.66mg/mL) for 24 and 48 hours were treated. In our study, 6.66mg/mL concentrations of entecavir in an increased number of cells with chromosomal abnormalities and structural chromosomal abnormalities in 48 hours of application and the increase was found to be significant when analyzed statistically. Additionally, the Mitotic Index (MI) has fallen from 24 and 48 hours practice and it was determined that this decline was greater in the 24-hour application. Further more entecavir on the micronucleus formation was determined as well as control show a different effect. In addition, the nuclear fission index of entecavir just 24 hours of application (NBU) in it was determined that significantly lowers. As a result [2-4], entecavir in high doses to cause genotoxicity of long-term use and entecavir administered first 24 hours cytotoxic effect that and the mitotic activity in the subsequent process of the cells was observed that continued division by winning again.
As per the results of Entecavir is a guanosine analogue with activity against hepatitis B virus (Ref: Celen MK, Dal T, Ayaz C, Bayan K, Mert D, Devecil O, Oruc EK). The aim of this 4-year trial was to evaluate entecavir treatment in nucleos (t) ide-naïve HBeAg-positive chronic hepatitis B patients. Forty-nine patients received entecavir and nine of them withdrew from the trial at the end of week 96. The initial mean value of alanine aminotransferase was 79.4+/-41.5 IU/L, and at the end of the 4-year study period, 90% of patients had alanine aminotransferase values within the normal range. At week 96, 91.7% of patients had HBV DNA<300 copies [5]; at month 48, 90% of patients had HBV DNA<50 IU/mL. HBeAg loss was recorded in 7.1% of patients at week 96 and in 12.5% at month 48. The rate of HBeAg seroconversion was 4.8% at week 96 and 7.5% at month 48. The rate of HBsAg seroconversion was 2.1% at week 96 and 2.5% at month 48. Entecavir as a potent and safe agent leading to continuous viral suppression proved to be safe and well tolerated therapy [6].
The initial patent on entecavir expired in South Africa in 2011 ZA 1991/07894. Current status available on: http://patentsearch.cipc.co.za/ [7] which should have permitted lower-cost generic competitors to enter the market. However, South Africa granted BMS three additional patents on entecavir that only expire between 2022 and 2026. Two of these patents have lapsed-meaning BMS has not paid the renewal fees, and they cannot be enforced-while one patent covering a lower dosage form of entecavir remains in force. This patent is currently under litigation in India Basheer S. BMS Hepatitis Patent Invalidated: A Viral Effect for India? http://spicyip.com/2013/02/bms-hepatitis-patent-invalidated-viral.html but because it is in force in South Africa, generic suppliers may be discouraged from bringing their low-dose products to market. A more recent patent on entecavir has not yet been received or processed by the patents office, but it could be filed up until the end of 2014 patent number: WO/2013/177672. Current status available on pa-tentscope.wipo.int. This patent covers the manufacturing process of entecavir, and is an example of patent evergreening-where companies file patents on minor changes to an existing drug to maintain patent protection and block competition [8]. The same patent was recently overturned in the United States for failing to meet the criteria of inventive step. However, in South Africa, since no examination of patent applications occurs, if the patent is filed, it is likely to be granted to BMS. So long as BMS maintains a monopoly on entecavir in South Africa, the price is likely to remain high, and entecavir will remain out of reach for those who need it. But the crystalline forms of entecavir and its performances are not researched and reported in the above-mentioned patent. Entecavir also helps to prevent the hepatitis B virus from multiplying and infecting new liver cells, is also indicated for the treatment of chronic hepatitis B in adults with HIV/AIDS infection [9-13].
To study the genotoxicity of entecavir, we evaluated the effects of entecavir on a panel of gene disrupted clones below table 1 by MTT assay. Camptothecin (CPT), a topoisomerase I poison, was selected as a positive control. We continuously exposed WT and mutant cells to entecavir or CPT at various concentrations for 72h [14-16]. The results indicated that entecavir inhibited the growth of DT40 cells in a dose-dependent manner. As shown in figure 1.
|
Gene |
Function |
References |
|
Rev3 |
TLS, HR (Catalytic submit of polξ) |
[17] |
|
XPA |
An initial step of nucleotide excision repair |
[18] |
|
UBC13 |
Ubc13 is related to the initial step of HR and postreplication repair |
[14,19] |
|
Parp1-/- |
Poly (ADP) ribosylation, related to single-strand break and base excision repair |
[15] |
|
Brca1 |
HR |
[16] |
|
Brca2 |
HR |
[20] |
|
Rad18 |
TLS |
[18] |
|
Polβ |
Base excision repair |
[21] |
|
Fen1 |
Base excision repair, processing 5' flap in long-patch and lagging stand DNA replication |
[22] |
|
Xrcc2 |
Rad51 paralog, homologous recombination, promotion of Rad51 assembly |
[23] |
|
Ctlp (S332A-/-) |
Eliminating covalently bound polypeptides from DSBs |
[24] |
|
Ku70 |
Intial Step for NHEJ dependent DSB repair |
[25] |
Table 1: Effects of entecavir on a panel of gene disrupted clones.
Note: DSB: Double-Starand Break; HR: Homologous Recombination; NHEJ: Nonhomologous End Joining Repair; TLS: Translesion DNA Synthesis.
 Figure 1: Mutant cells defective in DNA repair pathways were sensitive to entecavir.
Figure 1: Mutant cells defective in DNA repair pathways were sensitive to entecavir.
(A) The X-axis represents the concentration of entecavir and the Y-axis represents the relative number of surviving cells at 72 hours. Survival data were log-transformed giving approximate normality. Analysis of Covariance (ANCOVA) was used to test for differences in the linear dose-response curves between wild-type and a series of mutant cells. A p-value<0.05 was considered to be significant.
(B) Relative IC50 values of cell survival results in wild-type and their mutants exposed to entecavir or CPT. Each IC50 value was calculated from results of cell survival data shown in figure 1, [26-30], relative IC50 values were normalized according to the IC50 value of parental wild-typecells.
The IC50 [14-16,20,21] was calculated by SPSS software version 13.0. Data shown are the means of three experiments. Values shown are mean±SD, Parp1-/- -/- cells defective in DNA SSB exhibited the hypersensitivity to entecavir. Ubc13 deficient cells and TLS-deficient clones, both Rad18-/- and Rev3-/-, were sensitive to entecavir. To investigate the two major double-strand break repair pathways [31,32], HR and NHEJ, Brca1-/- , Brca2l-/-, Xrcc2-/- and Ku70-/- were analyzed. Only Brca1-/- cells manifested significant sensitivity to entecavir. Xrcc2-/- cells were even slightly resistant to entecavir [33]. The other DNA repair gene deficient cells, including XPA-/- cells were also sensitive to entecavir, but Polβ-/- , Fen1-/-and CtIP (S332A-/- ) cells were not. CPT can induce DNA damage by inhibiting the ligation of SSBs that are formed during the normal functioning of topoisomerase I. Unrepaired SSBs are converted to double-strand breaks upon replication. It has been shown that CPT induced double-strand breaks are mainly repaired by HR in DT40 cells. As shown in figure 2.
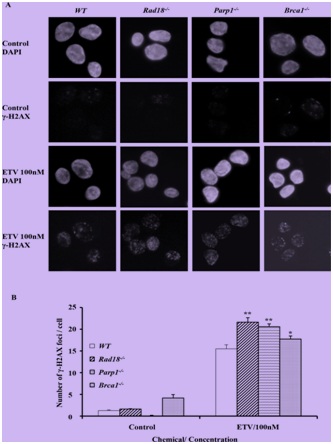 Figure 2: Entecavir induced the accumulation of γ-H2AX in nuclei of DT40 cells.
Figure 2: Entecavir induced the accumulation of γ-H2AX in nuclei of DT40 cells.
(A) Immuno-staining of Wild-Type (WT) and mutant DT40 clones using anti-γ-H2AX antibody and DAPI. Cells were fixed 6 hours after treated with entecavir 100nM. ETV, entecavir. (B) Quantification of γ-H2AX foci in individual cells of the indicated genotype. Cells were treated with entecavir 100nM for 6h. Data shown are the means of three experiments. Values shown are mean±SD. **P<0.01, *P
Parp1-/-, Rad18-/-, Ubc13-/-, CtIP(S332A-/-), Brca1-/- and Brca2l-/- cells were hypersensitive to CPT. In contrast, Polβ-/- and Ku70-/- were resistant to CPT, as previously reported. This observation indicated that entecavir may exert potential genotoxic mechanisms which mainly associate with SSB repair and PRR, but not a double-strand break repair.
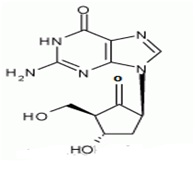
The chemical name for entecavir is 2-amino-I,9-dihydro-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl)-6H-purin-6-one, monohydrate. Its molecular formula is C12H15N5O3.H2O, which corresponds to a molecular weight of 295.3 Entecavir, BMS-200475, SQ-34676(1S,3R,4S)-9-[4-Hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl] guanine CAS-42217-69-4, 209216-23-9 (monohydrate).
DISCUSSION
Several methods have been developed for the determination of Entecavir and its enantiomers by HPLC, LCMS techniques [27,28]. Entecavir is an oral antiviral drug used in the treatment of Hepatitis B Virus (HBV) infection. Entecavir is a reverse transcriptase inhibitor 1 [11]. It prevents the hepatitis B virus from multiplying and reduces the amount of virus in the body. More specifically, it is a deoxyguanosine [34], analogue that inhibits reverse transcription, DNA replication and transcription in the viral replication process. Entecavir belonging to the chemical class of purine derivatives and chemically it is 2-amino-9-[(1S, 3R, 4S)-4-hydroxy-3-(hydroxymethyl)-2-methylidenecyclopentyl]-3H-purin-6-one with molecular formula C12H15N5O3.H2O3. Entecavir is a white to off-white powder [35]. It is slightly soluble in water (2.4mg/mL), and the pH of the saturated solution in water is 7.9 at 25°C±0.5°C. Baraclude film-coated tablets are available for oral administration in strengths of 0.5mg and 1mg of Entecavir.
Entecavir induced the accumulation of γ-H2AX in nuclei of DT40 cells
Patent information
Entecavir was successfully developed by Bristol-Myers Squibb Co. of USA first and the trademark of the product formulation is Baraclude™, including two types of formulations of tablet and oral solution having 0.5mg and 1mg of dosage. Chinese publication No. CN1310999 made by COLONNO, Richard J et al., discloses a low amount of entecavir and uses of the composition containing entecavir in combination with other pharmaceutically active substances for treating hepatitis B virus infection, however, the entecavir is non-crystal. In addition, its oral formulations such as tablet and capsule are made by a boiling granulating process [16,20,21]. The process is too complicated to control quality of products during humidity heat treatment even though ensuring uniform distribution of the active ingredients [14].
Synthesis of BMS-200475 (EN: 182634) The regioselective reaction of cyclopentadiene (I) and sodium (1) or commercial sodium cyclopentadienide (II) (2, 3) with benzyl chloromethyl ether (III) by means of the chiral catalyst (-)-diisopinocampheylborane in THF, followed by hydroxylation with H2O2/NaOH, gives (1S-trans)-2-(benzyloxymethyl)-3-cyclopenten-1-ol (IV), which is regioselectively epoxidized with tert-butyl hydroperoxide and vanadyl acetylacetonate in 2,2,4-trimethylpentane,yielding[1S-(1alpha,2alpha,3beta,5alpha)-2-(benzyloxymethyl)-6-oxabicyclo [3.1.0] hexan-3-ol (V). The protection of (V) with benzyl bromide and NaH affords the corresponding ether (VI), which is condensed with 6-O-benzylguanine (VII) by means of LiH in DMF to give the guanine derivative (VIII). The protection of the amino group of (VIII) with 4-methoxyphenyl(diphenyl)chloromethane (IX), TEA and DMAP in dichloromethane gives intermediate (X), which is oxidized at the free hydroxyl group with methylphosphonic acid, DCC and oxalic acid in DMSO (1) or Dess Martin periodinane in dichloromethane (2, 3), yielding the cyclopentanone derivative (XI). The reaction of (XI) with (i) Zn/TiCl4/CH2Br2 complex in THF/CH2Cl2 (1), (ii) activated Zn/PbCl2/CH2I2/TiCl4 in THF/CH2Cl2 (2), (iii) Nysted reagent/TiCl4 in THF/CH2Cl2 (2, 3) or (iv) Tebbe reagent in toluene (2) affords the corresponding methylene derivative (XII), which is partially deprotected with 3N HCl in hot THF, providing the dibenzylated compound (XI). Finally, this compound is treated with BCl3 in dichloromethane (1-3). (Scheme 18263401a) Description Hydrate, m.p>220C, alpha (22,D) +34? (c 0.3, water) (1); monohydrate, white crystalline solid, m.p. 234-6C (decomp.) for the bulk sample and m.p. 255C (decomp.) for an analytical sample recrystallized from water, alpha (D) +33.2? (c 0.3, water) (2); alpha (D) +35.0? (c 0.38, water) (3). Manufacturer Bristol-Myers Squibb Co. (US). References 1. Zahler R, Slusarchyk WA (Bristol-Myers Squibb Co.). Hydroxymethyl (methylenecyclopentyl) purines and pyrimidines. EP 481754, JP 92282373, US 5206244. 2. Bisacchi GS, Sundeen JE (Bristol-Myers Squibb Co.). Improved process for preparing the antiviral agent [1S-(1alpha, 3alpha, 4beta)]-2-amino-1,9-dihydro-9-[4-hydroxy-3-(hydroxymethyl)-2-methylenecyclopentyl]-6H-purin-6-one. WO 9809964. 3. Bisacchi GS, Chao ST, Bachard C, et al., BMS-200475, a novel carboxylic 2'-deoxyguanosine analog with potent and selective anti-hepatitis B virus activity in vitro. Bioorged Chem Lett 1997, 7: 127-32.EP 0481754; JP 1992282373; US 520624 4. Zahler R, Slusarchyk WA (Bristol-Myers Squibb Co.); Hydroxymethyl (methylenecyclopentyl) purines and pyrimidines. EP 0481754; JP 1992282373; US 5206244, EP 0481754; JP 1992282373; US 5206244; WO 9809964
DNA repair-deficient cells showed a marked increase in entecavir-induced chromosome breaks
 Entecavir-Anti-Hepatitis Virus Drugs, Antiinfective therapy, Antiviral drugs-Phase III
Entecavir-Anti-Hepatitis Virus Drugs, Antiinfective therapy, Antiviral drugs-Phase III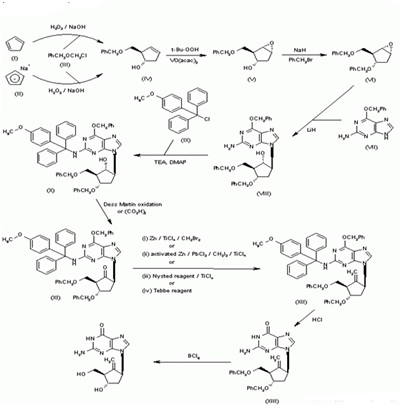 Entecavir-Route of synthesis.
Entecavir-Route of synthesis.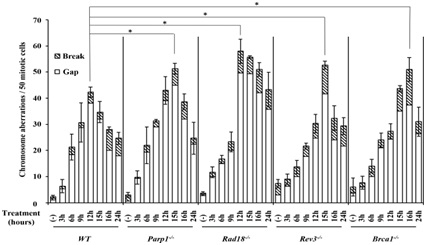 Figure 3: DNA repair-deficient cells showed a marked increase in entecavir-induced chromosome breaks.
Figure 3: DNA repair-deficient cells showed a marked increase in entecavir-induced chromosome breaks. Figure 4: Representative karyotype analysis of entecavir pretreated Rad18-/- cells.
Figure 4: Representative karyotype analysis of entecavir pretreated Rad18-/- cells.Increased frequency of Chromosomal Aberrations (CAs) in DNA repair-deficient cells and WT treated with entecavir (200nM) from 3 hours to 24 hours. Data are derived from 50 metaphase cells for each treatment. The experiments were independently repeated three times for statistical analysis. Values shown are mean±SD. *P<0.05 compared to WT. The differences between the WT and DNA [39,40,43], repair-deficient cell lines were tested for statistical significance using t-test.
Entecavir scale up process
SSBs in DNA are often raised by loss of a single nucleotide and by damaged 5’-and/or 3’-termini at the site of the break A multitude of factors trigger SSBs. Erroneous incorporation of ribonucleotides into DNA is the commonest sources of endogenous SSBs Parp1-/- is a sensor protein, which plays an important role in DNA SSB detection. In the current study, we found that Parp1-/- -/- cells exhibited the hypersensitive to entecavir and manifested significantly increase in the number of γ-H2AX foci and chromosomal aberrations compared with WT, suggesting that entecavir may induce SSBs. As Parp1-/- also functions in BER, we examined the sensitivity of BER deficient cells Polβ-/- , and results showed Polβ-/- cells were not significantly sensitive to entecavir. But we found the NER deficient cells XPA-/- were slightly sensitive to entecavir.
We also examined Brca1-/- , Brca2-/-, Xrcc2-/-, CtIP(S332A-/- ) and Ku70-/- cells [53], which respectively defective in HR and NHEJ, two major pathways for double strand breaks repair, and only Brca1-/- cells showed sensitivity to entecavir. We speculate that double strand breaks might not be the majority of entecavir-induced DNA damages. Recent studies had proved that besides the function on HR for double strand breaks repair, Brca1 could directly recruits translesion polymerases, such as Polη and Rev1, to the lesions through protein-protein interactions, suggesting its critical role in PRR [54-56]. Currently, we found Rev3-/- and Rad18-/- were also sensitive to entecavir and had increased entecavir-induced chromosomal aberrations (Figure 3). Both Rad18 and Rev3 play critical role in PRR pathway. Studies indicated that Rad18 forms a complex with Rad6 to promote PCNA mono-ubiquitination, which is a crucial step in PRR pathway, where as Rev3 gene encodes the catalytic subunit of DNA Polξ, which is involved in TLS, one of PRR pathway [54,57-59]. Furthermore, Ubc13, a K63-linked E2 Ub-conjugating enzyme, have been proved to function on both HR and error-free PRR. Results showed cells deficient in Ubc13 were also sensitive to entecavir. Above all, we hypothesize that entecavir induces DNA damage, which may collapse the replication forks and PRR pathway might release the replication fork stall (Figure 4).
(A) Representative karyotype of untreated Rad18-/- cells. (B) Chromosomal Aberrations (CAs) in Rad18-/-cells following 200nM entecavir pretreatment for 15h. Macrochromosomes 1-5 and Z are identified. Chromosome gaps are shown by arrow.
Entecavir was metabolized by phosphorylation to Triphosphate (TP) form in mammalian cells by cellular enzymes to inhibit HBV DNA replication [60]. The mechanism for chain termination by entecavir is likely to involve incorporation and abortive extension of ETV-containing HBV DNA. Some studies reported that entecavir displays no interaction with host polymerase and failed to be incorporated into human DNA. Nonetheless, the recent study by Brown et al., showed that entecavir can be incorporated and embedded into the human genome via primer extension with human X or Y polymerases or subsequent ligation. One possible model that could explain our data is shown in figure 5. The triphosphate of entecavir is incorporated into DNA strand by host replication or repair polymerases, which blocking extension of the nascent strand and inducing DNA, SSB and Parp1-/- dependent repair [61]. The entecavir-induced DNA lesions could also be repaired by PRR to avoid the replication fork collapse and chromosomal breaks when cells enter into S phase [36,62,63].
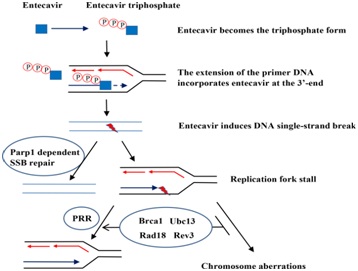 Figure 5: Model of entecavir-induced genotoxicity related to Single-Strand Break (SSB) repair and Postreplication Repair (PRR) pathway.
Figure 5: Model of entecavir-induced genotoxicity related to Single-Strand Break (SSB) repair and Postreplication Repair (PRR) pathway.The triphosphate of entecavir is incorporated into DNA strand by host replication or repair polymerases, which blocking extension of the nascent strand and inducing DNA SSB and Parp1-/- dependent repair. The entecavir-induced DNA lesions could also be repaired by PRR to avoid the replication fork collapse and chromosomal breaks when cells enter into S phase NAs have been shown effective inhibition of HBV replication, which delay the progression of liver cirrhosis, reduce the incidence of HBV related liver cancer, above all, increase the life span of the patients. Until now, most current guidelines recommended that a long-term treatment with NAs is essential to majority CHB patients, even a life-long therapy for CHB with cirrhosis [64]. And entecavir is one of the first-line therapies [65]. Especially in those with decompensated liver disease, undergoing immunosuppressive treatment or with contraindications, and those unwilling to receive Peg-IFN, entecavir or tenofovir is the only therapeutic options in patients. However, long-term safety data are still lacking for NAs, including entecavir [52,66-68], [Some studies demonstrated entecavir was clastogeic at 36μM in primary human lymphocytes]. Considering that entecavir inhibited HBV DNA synthesis in the nanomolar range, so they thought it’s safe to humans. But in our study, entecavir induced DNA damage at nanomolar in DT40 cells, especially in the more sensitive DNA repair deficient cells. So we think it is necessary to monitor the genotoxicity of NAs, especially entecavir, and to restrict treatment period (Figure 6).
CONCLUSION
Much work remains to be done to gain a better understanding of the mechanism of genotoxicity of entecavir. A better understanding of entecavir-induced genotoxicity may contribute to development of new drugs for the treatment or prevention of chronic hepatitis B with higher therapeutic efficacy and less genotoxicity. Some data already published evaluate the efficacy and safety of Entecavir (ETV) among Chronic Hepatitis B (CHB) nucleos (t) ide-naive Egyptian patients results ETV proved to have a potent antiviral efficacy and safety in nucleoside/tide-naive Egyptian patients. Rate of HBV DNA undetectability was higher in patients above 40 years of age and in patients who initially had a low viral load. ETV was well tolerated during the treatment period with a good overall safety profile Ref: https://www.ncbi.nlm.nih.gov/pmc/articles/Brca1-/-
REFERENCES
- World Health Organization (2017) Hepatitis B. World Health Organization, Geneva, Switzerland.
- Zhao GY, Sonoda E, Barber LJ, Oka H, Murakawa Y, et al. (2007) A critical role for the ubiquitin-conjugating enzyme Ubc13 in initiating homologous recombination. Mol Cell 25: 663-675.
- McKenna S, Spyracopoulos L, Moraes T, Pastushok L, Ptak C, et al. (2001) Noncovalent interaction between ubiquitin and the human DNA repair protein Mms2 is required for Ubc13-mediated polyubiquitination. J Biol Chem 276: 40120-40126.
- Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, et al. (1998) XRCC1 is specifically associated with poly (ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol 18: 3563-3571.
- Mazzucco CE, Hamatake RK, Colonno RJ, Tenney DJ (2008) Entecavir for treatment of hepatitis B virus displays no in vitro mitochondrial toxicity or DNA polymerase gamma inhibition. Antimicrob Agents Chemother 52: 598-605.
- Liaw YF, Raptopoulou-Gigi M, Cheinquer H, Sarin SK, Tanwandee T, et al. (2011) Efficacy and safety of entecavir versus adefovir in chronic hepatitis B patients with hepatic decompensation: A randomized, open-label study. Hepatology 54: 91-100.
- Chae HB, Kim JH, Kim JK, Yim HJ (2009) Current status of liver diseases in Korea: Hepatitis B. Korean J Hepatol 6: 13-24.
- Okada T, Sonoda E, Yamashita YM, Koyoshi S, Tateishi S, et al. (2002) Involvement of vertebrate polkappa in Rad18-independent postreplication repair of UV damage. J Biol Chem 277: 48690-48695.
- Korean Association for the Study of the Liver (2012) KASL clinical practice guidelines: Management of chronic hepatitis B. Clin Mol Hepatol 18: 109-162.
- Pol S, Lampertico P (2012) First-line treatment of chronic hepatitis B with entecavir or tenofovir in 'real-life' settings: From clinical trials to clinical practice. J Viral Hepat 19: 377-386.
- Chang TT, Gish RG, de Man R, Gadano A, Sollano J, et al. (2006) A comparison of entecavir and lamivudine for HBeAg-positive chronic hepatitis B. N Engl J Med 354: 1001-1010.
- Lai CL, Shouval D, Lok AS, Chang TT, Cheinquer H, et al. (2006) Entecavir versus lamivudine for patients with HBeAg-negative chronic hepatitis B. N Engl J Med 354: 1011-1020.
- Sherman M, Yurdaydin C, Sollano J, Silva M, Liaw YF, et al. (2006) Entecavir for treatment of lamivudine-refractory, HBeAg-positive chronic hepatitis B. Gastroenterology 130: 2039-2049.
- European Association For The Study Of The Liver (2012) EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J Hepatol 57: 167-185.
- Liaw YF, Leung N, Kao JH, Piratvisuth T, Gane E, et al. (2008) Asian-Pacific consensus statement on the management of chronic hepatitis B: A 2008 update. Hepatol Int 2: 263-283.
- Lok AS, McMahon BJ (2009) Chronic hepatitis B: Update 2009. Hepatology 50: 661-662.
- Santantonio TA, Fasano M (2014) Chronic hepatitis B: Advances in treatment. World J Hepatol 6: 284-292.
- Tenney DJ, Rose RE, Baldick CJ, Pokornowski KA, Eggers BJ, et al. (2009) Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 49: 1503-1514.
- Yang SW, Kim GH, Chung JW, Sohn HR, Lee SS, et al. (2015) Prediction of risk for hepatocellular carcinoma by response of serum α-fetoprotein to entecavir therapy. J Gastroenterol Hepatol 30: 1175-1182.
- Brambilla G, Mattioli F, Robbiano L, Martelli A (2012) Studies on genotoxicity and carcinogenicity of antibacterial, antiviral, antimalarial and antifungal drugs. Mutagenesis 27: 387-413.
- Tenney DJ, Rose RE, Baldick CJ, Pokornowski KA, Eggers BJ, et al. (2009) Long-term monitoring shows hepatitis B virus resistance to entecavir in nucleoside-naive patients is rare through 5 years of therapy. Hepatology 49: 1503-1514.
- Dalmora SL, Sangoi Mda S, Nogueira DR, da Silva LM (2010) Validation of a stability-indicating RP-HPLC method for the determination of entecavir in tablet dosage form. J AOAC Int 93: 523-530.
- Amirtharaj VR, Kumar VC, Kumar SN (2011) Development and validation of RP-HPLC for the estimation of entecavir in tablet dosage form. Int J Res Pharm Biomed Sci 2: 1033-1040.
- Rambabu R, Subbarao J, Vidyadhara S (2014) Estimation and validation of entecavir in bulk and pharmaceutical dosage forms by RP-HPLC. Int J Res Ayurveda Pharm 5: 531-535.
- Asaad AM, Al-Ayed MS, Aleraky M, Qureshi MA (2015) Hepatitis B virus genotyping in chronic hepatitis B patients in southwestern Saudi Arabia. Braz J Infect Dis 19: 525-528.
- Brusky J, Zhu Y, Xiao W (2000) UBC13, a DNA-damage-inducible gene, is a member of the error-free postreplication repair pathway in Saccharomyces cerevisiae. Curr Genet 37: 168-174.
- Friedrich A, Olejniczak K (2011) Evaluation of carcinogenicity studies of medicinal products for human use authorised via the European centralised procedure (1995-2009). Regul Toxicol Pharmacol 60: 225-248.
- http://www.baraclude.bmscustomerconnect.com/
- Martin P, Lau DT, Nguyen MH, Janssen HL, Dieterich DT, et al. (2015) A Treatment algorithm for the management of chronic hepatitis B virus infection in the United States: 2015 update. Clin Gastroenterol Hepatol 13: 2071-2087.
- Brown JA, Pack LR, Fowler JD, Suo Z (2012) Presteady state kinetic investigation of the incorporation of anti-hepatitis B nucleotide analogues catalyzed by noncanonical human DNA polymerases. Chem Res Toxicol 25: 225-233.
- Yamamoto KN, Hirota K, Kono K, Takeda S, Sakamuru S, et al. (2011) Characterization of environmental chemicals with potential for DNA damage using isogenic DNA repair-deficient chicken DT40 cell lines. Environ Mol Mutagen 52: 547-561.
- Buerstedde JM, Takeda S (1991) Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell 67: 179-188.
- Yamazoe M, Sonoda E, Hochegger H, Takeda S (2004) Reverse genetic studies of the DNA damage response in the chicken B lymphocyte line DT40. DNA Repair (Amst) 3: 1175-1185.
- Tenney DJ, Pokornowski KA, Rose RE, Baldick CJ, Eggers BJ, et al. (2009) 20 Entecavir maintains a high genetic barrier to HBV resistance through 6 years in naive patients. J Hepatol 50: 10.
- Shim JH, Lee HC, Kim KM, Lim YS, Chung YH, et al. (2010) Efficacy of entecavir in treatment-naïve patients with hepatitis B virus-related decompensated cirrhosis. J Hepatol 52: 176-182.
- Evans TJ, Yamamoto KN, Hirota K, Takeda S (2010) Mutant cells defective in DNA repair pathways provide a sensitive high-throughput assay for genotoxicity. DNA Repair (Amst) 9: 1292-1298.
- Zoutendijk R, Reijnders JG, Brown A, Zoulim F, Mutimer D, et al. (2011) Entecavir treatment for chronic hepatitis B: Adaptation is not needed for the majority of naïve patients with a partial virological response. Hepatology 54: 443-451.
- Fung J, Seto WK, Lai CL,Yuen MF (2014) Extrahepatic effects of nucleoside and nucleotide analogues in chronic hepatitis B treatment. J Gastroenterol Hepatol 29: 428-434.
- Hu J, Nakamura J, Richardson SD, Aitken MD (2012) Evaluating the effects of bioremediation on genotoxicity of polycyclic aromatic hydrocarbon-contaminated soil using genetically engineered, higher eukaryotic cell lines. Environmental Science & Technology 46: 4607-4613.
- Sonoda E, Okada T, Zhao GY, Tateishi S, Araki K, et al. (2003) Multiple roles of Rev3, the catalytic subunit of polzeta in maintaining genome stability in vertebrates. EMBO J 22: 3188-3197.
- Wu X, Takenaka K, Sonoda E, Hochegger H, Kawanishi S, et al. (2006) Critical roles for polymerase zeta in cellular tolerance to nitric oxide-induced DNA damage. Cancer Res 66: 748-754.
- Mizutani A (2004) Extensive chromosomal breaks are induced by tamoxifen and estrogen in DNA repair-deficient cells. Cancer Res 64: 3144-3147.
- Danoy P, Sonoda E, Lathrop M, Takeda S, Matsuda F (2007) A naturally occurring genetic variant of human XRCC2 (R188H) confers increased resistance to cisplatin-induced DNA damage. Biochem Biophys Res Commun 352: 763-768.
- Korea Centers for Disease Control & Prevention (2012) Korea National Health and Nutrition Examination Survey. Korea Centers for Disease Control & Prevention, Cheongju, South Korea.
- Bae SH, Yoon SK, Jang JW, Kim CW, Nam SW, et al. (2005) Hepatitis B virus genotype C prevails among chronic carriers of the virus in Korea. J Korean Med Sci 20: 816-820.
- Kim H, Jee YM, Song BC, Shin JW, Yang SH, et al. (2007) Molecular epidemiology of Hepatitis B Virus (HBV) genotypes and serotypes in patients with chronic HBV infection in Korea. Intervirology 50: 52-57.
- Bristol-Myers Squibb Company (2017) Antiviral Drugs Advisory Committee (AVDAC) briefing document. Bristol-Myers Squibb Company, New York, USA.
- National Institutes of Health (2017) Study of entecavir in patients with chronic hepatitis B virus infection. National Institutes of Health, Maryland, USA.
- Hou JL, Jia JD, Wei L, Zhao W, Wang YM, et al. (2013) Efficacy and safety of entecavir treatment in a heterogeneous CHB population from a ‘real-world’ clinical practice setting in China. J Viral Hepat 20: 811-820.
- Smirnova M, Klein HL (2003) Role of the error-free damage bypass postreplication repair pathway in the maintenance of genomic stability. Mutat Res 532: 117-135.
- Tian F, Sharma S, Zou J, Lin SY, Wang B, et al (2013) BRCA1 promotes the ubiquitination of PCNA and recruitment of translesion polymerases in response to replication blockade. Proc Natl Acad Sci U S A 110: 13558-13563.
- Manns MP, Akarca US, Chang TT, Sievert W, Yoon SK, et al. (2012) Long-term safety and tolerability of entecavir in patients with chronic hepatitis B in the rollover study ETV-901. Expert Opin Drug Saf 11: 361-368.
- Innaimo SF, Seifer M, Bisacchi GS, Standring DN, Zahler R, et al. (1997) Identification of BMS-200475 as a potent and selective inhibitor of hepatitis B virus. Antimicrob Agents Chemother 41: 1444-1448.
- Matsuzaki Y, Adachi N, Koyama H (2002) Vertebrate cells lacking FEN-1 endonuclease are viable but hypersensitive to methylating agents and H2O2. Nucleic Acids Res 30: 3273-3277.
- Nakamura K, Kogame T, Oshiumi H, Shinohara A, Sumitomo Y, et al. (2010) Collaborative action of Brca1 and CtIP in elimination of covalent modifications from double-strand breaks to facilitate subsequent break repair. PLoS Genet 6: 1000828.
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, et al. (1998) Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 17: 5497-5508.
- Qing Y, Yamazoe M, Hirota K, Dejsuphong D, Sakai W, et al. (2011) The epistatic relationship between BRCA2 and the other RAD51 mediators in homologous recombination. PLoS Genet 7: 1002148.
- Hatanaka A, Yamazoe M, Sale JE, Takata M, Yamamoto K, et al. (2005) Similar effects of Brca2 truncation and Rad51 paralog deficiency on immunoglobulin V gene diversification in DT40 cells support an early role for Rad51 paralogs in homologous recombination. Mol Cell Biol 25: 1124-1134.
- Prasad R, Williams JG, Hou EW, Wilson SH (2012) Pol ß associated complex and base excision repair factors in mouse fibroblasts. Nucleic Acids Res 40: 11571-1182.
- Hsiang YH, Hertzberg R, Hecht S, Liu LF (1985) Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem 260: 14873-14878.
- Adachi N, So S, Koyama H (2004) Loss of nonhomologous end joining confers camptothecin resistance in DT40 cells. Implications for the repair of topoisomerase I-mediated DNA damage. J Biol Chem 279: 37343-37348.
- Hochegger H, Sonoda E, Takeda S (2004) Post-replication repair in DT40 cells: Translesion polymerases versus recombinases. Bioessays 26: 151-158.
- Broomfield S, Hryciw T, Xiao W (2001) DNA postreplication repair and mutagenesis in saccharomyces cerevisiae. Mutat Res 486: 167-184.
- Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, et al. (2003) Histone H2AX phosphorylation is dispensable for the initial recognition of DNA breaks. Nat Cell Biol 5: 675-679.
- Mazzucco CE, Hamatake RK, Colonno RJ, Tenney DJ (2008) Entecavir for treatment of hepatitis B virus displays no in vitro mitochondrial toxicity or DNA polymerase gamma inhibition. Antimicrob Agents Chemother 52: 598-605.
- Caldecott KW (2014) DNA single-strand break repair. Exp Cell Res 329: 2-8.
- Caldecott KW (2014) Molecular biology. Ribose--an internal threat to DNA. Science 343: 260-261.
- Godon C, Cordelieres FP, Biard D, Giocanti N, Megnin-Chanet F, et al. (2008) PARP inhibition versus PARP-1 silencing: Different outcomes in terms of single-strand break repair and radiation susceptibility. Nucleic Acids Res 36: 4454-4464.
Citation: Pathy K (2018) Entecavir Patent Evaluation & Genotoxicity. J Toxicol Cur Res 2: 005.
Copyright: © 2018 Krishnasarma Pathy, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.