Evaluation of the Local Neurotoxicity of Tripropylene Glycol Diacrylate, a Leachable from Ink
*Corresponding Author(s):
Melanie K BotheDepartment Of Toxicology, Fresenius Kabi Deutschland GmbH, Else-Kröner-Strasse 1, 61352 Bad Homburg, Germany
Tel:+49 61726866768,
Email:Melanie.Bothe@fresenius-kabi.com
Abstract
The local neurotoxicity of drugs administered peri- or epidurally can be influenced, among other factors, by leachables resulting from the packaging material. In this study, the neurotoxicity of Tripropylene Glycol Diacrylate (TPGDA), a leachable resulting from printing ink, was assessed in vitro and in vivo. Reduction of neuronal cell viability, as measured by the Neutral Red Uptake assay, occurred at concentrations of 12.5µM TPGDA or higher. Reduction of metabolic activity of neuronal cells, determined by the CellTiter-Glo® assay, started at concentrations of 6.25µM TPGDA. Changes in the spike and burst pattern, as measured by the eCiphr®Neuro assay, were observed with 1hr incubation of cells with concentrations of 20µM TPGDA. No histopathological changes in morphology of the spinal cord were observed after intrathecal administration of up to 0.65µM TPGDA. The concentration of 0.65µM TPGDA is thus the highest determined concentration without local neurotoxic effects in vivo in rats and thereby considered safe for administration to patients.
Keywords
Cytotoxicity; Intrathecal; Leachable; Neurotoxicity; Tripropylene glycol diacrylate
INTRODUCTION
During drug development, adverse effects of a drug product are assessed through numerous toxicological studies. The safety of a drug product, however, is not only dependent on the effects of the active pharmaceutical ingredient. Drug degradants, impurities, as well asleachables resulting from different components of the packaging material have a substantial impact as well, and their safety needs to be demonstrated. In this context, neuronal tissuesare of utmost importance due to their role in the central functions they control, and their limited regenerative capabilities. Impurities and leachables associated with drugs indicated for administration in close proximity to neuronal tissues, such asperidural local anesthetics, must therefore beevaluated for local neurotoxic effects.
Here, we investigated the local neurotoxic effect of Tripropyleneglycol Diacrylate (TPGDA, CAS 42978-66-5), a leachable resulting from printing ink (Product Quality Research Institute (PQRI))[1]. The systemic toxicity of TPGDA is well described. It is severely irritating to the eyes and classified as a skin sensitizer (European Chemicals Agency (ECHA))[2]. Data on skin irritation range from none to slight edema after 48h as well as slight to well define erythema after 72h of exposure (European Chemicals Agency (ECHA))[2]. Following oral administration, TPGDA induces adverse effects in the liver, but has not been shown to cause neurotoxic effects at doses up to 750mg/kg/day (European Chemicals Agency (ECHA))[2]. Local effects after oral administration are due to the irritating nature of the substance, and include hyperplasia and hyperkeratosis in the non-glandular portion of the stomach (European Chemicals Agency (ECHA))[2]. However, local neurotoxic effects of this irritating substancehave neither been determined in animals, nor in human beings. The aim of this study was to determine a concentration of TPGDA not eliciting local neurotoxic effects when administered in close proximity to neural tissue.
MATERIALS AND METHODS
Chemicals
TPGDA was obtained from Chemical Point UG (Deisenhofen, Germany).
In Vitro Experiments
Cell treatment for Neutral Red Uptake (NRU) and CellTiter-Glo® assay: The cells used in these assays werederived from a human bone marrow-derived neuroblastoma cell line, SHSY5Y, which is often used as an in vitro model for neuronal function and differentiation. Thecells were routinely grown in Ham’s DMEM/F-12 medium supplemented with 10% Fetal Bovine Serum at 37°C in a humidified atmosphere containing 5% carbon dioxide. One day prior to the experiments, cells were plated in 384-well tissue culture plates, 25μL/well at approximately 1x105cells/mL for CellTiter-Glo® assay, and 25µL/well at approximately 4x105cells/mL for NRU assay followed by overnight incubation. Subsequently, cells were treated with TPGDA (triplicate, 10 concentrations in 2-fold dilutions)or controls (duplicate, 10 concentrations in 2-fold dilutions and incubated at 37°C in a humidified atmosphere, 5% CO2 for 48hr. Chlorpromazine (CPZ) and carbonyl cyanide 3-Chlorophenylthydrazone (CCCP) were used as positive controls and Dimethyl Sulfoxide (DMSO) served as the vehicle control (0.5% final concentration)on all plates.
Neutral Red Uptake: Following 48hr incubation, media was removed and replaced with 25μL of prepared Neutral Red (33μg/mL NR) solution followed by further incubation at 37°C for 3hr. After incubation, Neutral Red solution was removed, cells were washed once with 25μL PBS and 50μL of Neutral Red Assay Solubilization Solution (49 parts water + 50 parts ethanol + 1 part acetic acid) was added to each well. The plate was placed on the plate shaker for 10min to fullyextract Neutral Red from the cells followed by absorption measurement via spectrophotometer at 540nm.
CellTiter-Glo®: After incubation with test compounds, 25µL media from each well was removed and replaced by 25μL of the CellTiter-Glo® (1:1 ratio). The plate was placedon a plate shaker for 2min and then incubated at room temperature in the dark for 10min. Luminescence was measured on a Synergy H1 plate reader within 20min.
Cell treatment for eCiphr®Neuro: Functional neurotoxicity was assessed with the eCiphr®Neuro assay. This assay used spike (single action potential) and burst (periods of rapid action potential spiking followed by quiescent periods) data from cryogenically preserved rat cortical neurons and to investigate the effect of compounds on several aspects of a spike pattern. 48-well microelectrode array plates were coated with polyethyleneimine, rinsed and allowed to dry in a sterile environment overnight at room temperature.
1hr prior to plating, a laminin solution was applied to the center of the well covering the electrodes and incubated at 37°C. Cryopreserved rat cortical neurons were thawed and slowly diluted with neurobasal medium supplemented with Gibco B-27 neuronal cell culture supplement, L-glutamine and penicillin-streptomycin (NB/B27) in a drop-wise manner to avoid osmotic shock. Following a centrifugation step, the supernatant was decanted, and the cell pellet was suspended in NB/B27 medium at a concentration of 5 x 106 cells per mL.
Prior to plating, the laminin solution was aspirated from the wells and the cells were plated onto the Microelectrode Array (MEA) plates resulting in 75,000 cells per well. The plates were incubated for 2hr at 37°C to allow sufficient attachment and then 250μL of NB/B27 medium was slowly added to each well. After a 1-day incubation period, an additional 250μL of the same medium was added to achieve a final well volume of 500μL. The cells were maintained in a humidified incubator at 37°C for 13-16 days with media changes 3 times a week before experimental procedures were performed. The activity of cells was recorded prior to dosing (baseline) using the Axion Biosystems Maestro microelectrode array system.
For dosing, 250μL of medium was removed from each well, dispensed into the corresponding wells of a sterile 48-well plate and mixed with vehicle, positive controls, or test compounds (DMSO final concentration 0.2%) at four concentrations in triplicate. The formulations were carefully added back to the corresponding wells of the MEA and incubated for 1hr at 37°C. A post-dose recording was obtained on the MEA system. Picrotoxin 10μM and Domoic acid 10μM were included as positive controls on all plates, while DMSO 0.2% was added as the negative control.
eCiphr®Neuro microelectrode array: All recordings were obtained with Axion Biosystems Maestro microelectrode array system (Axion Biosystems), a 768-channel high throughput MEA platform, utilizing 48-well plates configured with 16 electrodes per well. Prior to compound addition, a baseline of spontaneous spike activity was recorded using the Maestro system. At a predetermined time (one hourpost dose), an additional recording was obtained. Both raw data files were re-recorded using AxIS software’s Spontaneous Neural Configuration to generate a spike file.
Prior to baseline recordings, an assessment of activity was performed for each well of the 48-well MEA by observing spontaneous spike activity on the Maestro system. Wells with no or sparse activity were eliminated from the experiment determined by visual inspection of the live spike trains for each well.
After a 3-min equilibration time, baseline recordings of approximately 15min in length were obtained immediately before the addition of treatment compounds and controls. For analysis, electrodes with 100 or more spikes (~7 spikes/min) were determined to be “active” and only wells with 5 or more active electrodes were used in the final analysis. Following 1hr incubation at 37°C with compounds, another 15min recording was obtained after a 3min equilibration time. If a treated well fell below the activity threshold due to compound effect, only spike count was determined and reported. All other parameters were not calculated.
The following endpoints, calculated using custom MATLAB (MathWorks, Natick, MA) scripts, were reported:
• Firing Rate: Number of spikes normalized by time of the recording
• Burst Rate: Number of bursts normalized by time of the recording
• Number of Spikes in Burst: Calculated number of spikes that occur within bursts
• Percent of Isolated Spikes: Percent of spikes occurring outside of bursts
• Coefficient of Variation (CV) of the Inter Spike Intervals (ISI): Difference in time between adjacent spikes in each channel was computed to obtain the inter-spike intervals. The mean and standard deviation of the ISI’s for each channel was computed to yield a coefficient of variation
• Normalized IQR Burst Duration: Interquartile Range of the burst duration is normalized by the median of the burst duration
• Burst Duration: Length of time that a burst lasts between the first and last spike
• Inter burst Interval: Time between the trailing spike of each burst and the leading spike of the subsequent burst
• Inter spike interval distance: ISI-distance is calculated by the Kreuz method for spike train synchrony
• Normalized Mean Absolute Deviation (MAD) burst spike number: Statistical dispersion of the spikes in bursts
• Median ISI/Mean ISI: Median inter spike interval divided by the mean inter spike interval
• Median ISI: Median inter spike interval
Analysis of in vitro experiments: For the CellTiter-Glo®, the percent change relative to controls was calculated by dividing the luminescence value of each well of treated cells by the average luminescence value of the vehicle control wells and multiplying by 100. For the NRU assay, the percent change relative to controls was calculated by dividing the absorbance value of each well of treated cells by the average absorbance value of the vehicle control wells and multiplying by 100.
Response (% control) = [Response with Compound]/[Vehicle Response] x 100
A dose response curve was used to determine the IC50 using GraphPad Prism software for the results of the NRU and the CellTiter-Glo® assay. The percentages of control of each endpoint of the eCiphr®Neuro were statistically analysed with a two-way ANOVA.
In Vivo Experiment
Animals: A total of 60 Sprague Dawley rats, 30 males and 30 females, aged 9 to 10 weeks old and with an initial bodyweight of 351 to 406g (males) and 250 to 305g (females) were used in this study. Animals were group housed (up to 3 animals of the same sex and same dosing group together) in polycarbonate cages containing appropriate bedding equipped with an automatic watering valve. Target temperatures of 19°C to 25°C with a relative target humidity of 30% to 70% were maintained. A 12hr light/12hr dark cycle was maintained as well. PMI Nutrition International Certified Rodent Chow No. 5CR4 as well as tap water was provided ad libitum throughout the study. All animal experiments comply with the Canadian Council on Animal Care and are in accordance with the National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals (National Institute of Health) [3].
Experimental design: Animals were divided into 3 groups. Ten animals per sex of each group received either the vehicle or a low or a high dose of TPGDA via intrathecal injection. On day 2 after dosing, 5 animals per sex per group were euthanized and analyzed histopathologically. After a recovery period of 14 days, the remaining 5 animals per sex per group were euthanized and analyzed on day 15 after dosing.
Dosing: Animals of the vehicle control group received 50µL 0.9% sodium chloride for injection (Reference Item). Animals receiving TPGDA were administered 50µL of a 39 µg/L or 195 µg/L TPGDA containing 0.9% sodium chloride solution, resulting in dosages of 1.95ng/animal and 9.75ng/animal. The Test and Reference Items were administered once via a single direct intrathecal injection at the lumbar level (L4/L5). The dosing syringe was filled with 50μL of Test or Reference Item dose formulations and 0.2 to 0.3mL of air was attached to the needle and the volume was slowly injected. The wound was closed using a subcuticular suture of absorbable suture material.
The needle (with syringe) remained in the intrathecal space for at least 30 seconds aftercompletion of the injection. The position of the needle was confirmed so that the opening was facing rostral prior to the dose administration.
Observations: The animals were removed from the cage, and a detailed clinical observation was performed once pretreatment (on Day -1), weekly thereafter, and on the day of termination.
Tissue preparation: Main study and recovery animals surviving until scheduled euthanasia were weighed andeuthanized by exsanguination by incision from the abdominal aorta following isofluraneanesthesia. Main study and recovery animals were subjected to a complete necropsy examination, which included evaluation of the carcass and musculoskeletal system; all external surfaces and orifices; cranial cavity and external surfaces of the brain; and thoracic, abdominal, and pelvic cavities with their associated organs and tissues. Representative samples of the tissues were collected from all animals, preserved in 10% neutral buffered formalin, embedded in paraffin, sectioned, mounted on glass slides, andstained with hematoxylin and eosin for histopathological analysis.
Statistical analyses of in vivo experiments: Levene’s test was used to assess the homogeneity of group variances parametric assumption at the 5% significance level. Datasets with at least three groups were compared using an overall one-way ANOVA F-test or Kruskal-Wallis test (when parametric assumptions were not met) at the 5% significance level. The above pairwise comparisons were conducted using a two-sided Dunnett’s or Dunn’s test, respectively, when the overall test was significant. Datasets with two groups were compared using a two-sided t-test or Wilcoxon Rank-Sum test, respectively. All significant pairwise comparisons were reported at the 0.1, 1, and 5% significance levels.
RESULTS AND DISCUSSION
To investigate the local neurotoxic effects of TPGDA we undertook three in vitro experiments and one in vivo experiment in rats. In all in vitro experiments, the positive controls reacted as expected and were therefore considered valid. In the NRU assay, an experiment assessing the percentage of viable cells compared to the negative control, TPGDA revealed an IC50 of 22.35µM with the dose range of 0.195 - 100µM. The highest non-toxic concentration was 6.25µM (Figure 1). In the CellTiter-Glo® assay, an experiment determining the percentage of ATP produced by the metabolically active cells compared to the negative control, TPGDA revealed an IC50 of 12.92µM with the dose range of 0.195 - 100µM. The highest non-toxic concentration was 3.13µM (Figure 2). In the eCiphr®Neuro, no changes in neuronal activity occurred up to a concentration of 6.25µM TPGDA. At 20µM, TPGDA caused a decrease in the median number of spikes in burst, which was close to, but did not reach significance (p = 0.06).
In addition, 20µM TPGDA treatment of cells resulted in a statistically significant increase in percent isolated spikes.Statistically significant increases in the median Inter Spike Interval (ISI) as well as the median/mean ISI occurred with 2 and 20µM TPGDA (Figure 3). All other parameters were not statistically significantly different at 20µM concentration. The high variability of the median ISI in this very small number of samples (n=3) puts into question the biological relevance of the observed change in these two related endpoints (median ISI and median/mean ISI).
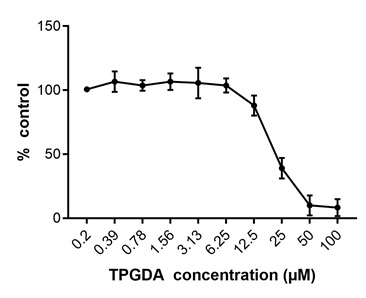 Figure 1: Neutral red uptake assay after 48hr incubation of neuronal cells with TPGDA. At concentrations of > 6.25µM, the percentage of viable cells decreased and was close to zero at concentrations > 50µM TPGDA. Data are given as mean ± SD of three individual wells.
Figure 1: Neutral red uptake assay after 48hr incubation of neuronal cells with TPGDA. At concentrations of > 6.25µM, the percentage of viable cells decreased and was close to zero at concentrations > 50µM TPGDA. Data are given as mean ± SD of three individual wells.
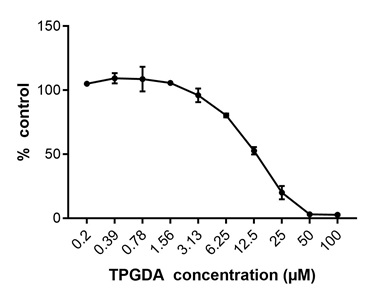 Figure 2: Cell Titer-Glo® assay after 48hr incubation of neuronal cells with TPGDA. At concentrations of > 3.13µM, the percentage of metabolically active cells decreased and was close to zero at concentrations > 50µM TPGDA. Data are given as mean ± SD of three individual wells.
Figure 2: Cell Titer-Glo® assay after 48hr incubation of neuronal cells with TPGDA. At concentrations of > 3.13µM, the percentage of metabolically active cells decreased and was close to zero at concentrations > 50µM TPGDA. Data are given as mean ± SD of three individual wells.
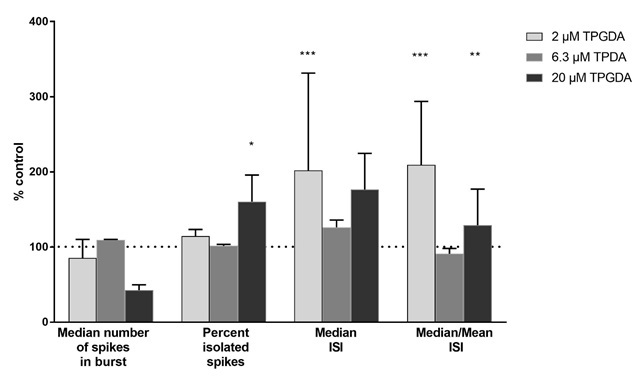 Figure 3: eCiphr®Neuro assay after 48hr incubation of neuronal cells with TPGDA. At concentrations 20µM, the median number of spikes in burst decreased, while the percentage of isolated spikes increased. Median Inter Spike Interval (ISI) and median/mean ISI both increased at concentration of 2 and 20µM, but not 6.3µM TPGDA. Data are given as mean ± SD of three individual wells. Dashed horizontal line = 100 % of mean control cell value.
Figure 3: eCiphr®Neuro assay after 48hr incubation of neuronal cells with TPGDA. At concentrations 20µM, the median number of spikes in burst decreased, while the percentage of isolated spikes increased. Median Inter Spike Interval (ISI) and median/mean ISI both increased at concentration of 2 and 20µM, but not 6.3µM TPGDA. Data are given as mean ± SD of three individual wells. Dashed horizontal line = 100 % of mean control cell value.
The in vitro cytotoxicity of TPGDA was also assessed in cell types other than neuronal cells by another research group. In normal human epidermal keratinocytes and bronchial epithelium,treatment with up to 200µM TPGDA did not induce cytotoxic effects, while normal human dermal fibroblasts only tolerated TPGDA concentrations of up to 40µM without cytotoxic effects [4]. Of note, incubation times were shorter in the experiments reported by Nylander-French and coworkers, 18hr instead of 48hr, and thus no direct comparison to our study can be done.
No TPGDA-related microscopic findings were noted at completion of the main or recovery periods after intrathecal injection of up to 195µg/L TPGDA (0.65µM) in rats. Furthermore, no TPGDA-related clinical observations or unscheduled deaths were observed throughout the study. One limitation of the study is that only concentrations of up to 0.65µM TPGDA were administered intrathecally. The actual concentrations without neurotoxic effects may thus be higher.
In summary, the main finding of this study is the determination of a non-neurotoxic concentration of 0.65µM TPGDA in vivo despite the irritant potential of this substance. Concentrations up to 3.13µM were tolerated for 48hr incubation in neuronal tissue in vitro and warrant further investigations of the actual highest non-neurotoxic dose in vivo. For the time being, 0.65µM TPGDA are considered safe for administration to human patients even in close proximity to vulnerable neuronal tissue.
ACKNOWLEDGEMENTS
We thank the following employees of Charles River for their participation in the study: Luc Chouinard for histopathological examinationand Nikita Navalkar for formulation analysis of the Test Item.
CONFLICT OF INTEREST
MKB, DF, and MW are employees of Fresenius Kabi Deutschland GmbH, the sponsor of this study.
STATEMENT OF AUTHORSHIP
MKB, DF, and MW designed the study and interpreted the data. ASDF and KM conducted the animal experiments and interpreted the data. SL, CS, and JB performed the in vitro experiments and interpreted the data. MKB and DF drafted the article and ASDF, KM, SL, CS, JB, and MW revised it critically and finally approved the version to be submitted.
REFERENCES
- PQRI (2011) Experimental protocol for qualitative controlled extraction studies on material test articles representative of Prefilled Syringe (PFS) and Small Volume Parenteral (SVP) container closure systems. Product Quality Research Institute, USA.
- ECHA (2020) Registration Dossier of CAS 15625-89-5. European Chemicals Agency, Helsinki, Finland.
- NIH (2011) Guide for the care and use of laboratory animals. In: The national academies collection. National Institutes of Health, Washington, D.C., USA.
- Nylander-French LA, French JE (2000) Comparative in vitro cytotoxicity of ethyl acrylate and tripropylene glycol diacrylate to normal human skin and lung cells. In vitro cellular & developmental biology-Animal 36: 611-616.
Citation: Bothe MK, Franckenstein D, Strock C, Bradley J, Di Fruscia AS, et al. (2020) Evaluation of the Local Neurotoxicity of Tripropylene Glycol Diacrylate, a Leachable from Ink. J Toxicol Cur Res 4: 017.
Copyright: © 2020 Melanie K Bothe, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.