Genetic Diagnosis of Fanconi Anemia
*Corresponding Author(s):
Selenti NDepartment Of Medical Genetics, Athens University School Of Medicine, Athens, Greece
Tel:+30 2107467462,
Email:nikoletta_selenti@hotmail.com
Abstract
Fanconi Anemia (FA) is a rare genetic disease characterized by congenital and developmental abnormalities, hypersensitivity to DNA cross-linking agents and progressive bone marrow failure.
Sixteen subtypes are currently recognized but deletions of the Fanconi Anemia Complementation Group A (FANCA) gene account for more than 65% of FA cases. This is a review of the genetic, molecular and clinical aspects of FA as well as of the future prospects.
Keywords
INTRODUCTION
Fanconi Anemia (FA) is a rare genetic disorder that is mainly inherited in an autosomal recessive pattern and is rarely X-linked. It was first described in 1927 by the Swiss pediatrician Guido Fanconi who described three affected siblings with congenital abnormalities and progressive marrow failure at the age of 4-5 years [1].
It is the most frequent inherited instability syndrome, characterized by bone marrow failure, hypersensitivity to cross-linking agents and high risk for Acute Myeloid Leukemia (AML). Ataxia Telangiectasia, Xeroderma pigmentosum and Bloom, Werner, Nijmegen, Li -Fraumeni and Seckel syndromes are also associated with inability to repair DNA damage [2,3].
Age of diagnosis ranges from birth to 49 years and the male-to-female ratio is 1.2:1, with a median age of 6,5 years in males and 8 years in females. The incidence rate is about 1 in 131,000 births in the USA; however the frequency is higher among specific populations due to founder effects [4].
FA patients exhibit hypersensitivity to agents that cause Interstrand DNA Cross-Links (ICLs), such as Mitomycin C (MMC), Diepoxybutane (DEB) and cisplatin, manifested as chromosomal fragility and radial formation that represent the biological hallmark of the disease. Chromosome instability is caused by defects in FA proteins which participate in several DNA repair pathways, including homologous recombination, DNA mismatch repair, nucleotide excision repair and translesion DNA synthesis [5,6].
CLINICAL FEATURES OF FA
Clinical features of FA patients include various congenital abnormalities, mainly skin and upper limbs disorders, (Table 1), progressive bone marrow failure and increased risk for squamous cell cancer [7-12]. However in 25-40% of patients the classical features may be very subtle or even absent [4,12,13].
| Organ system | Incidence | Abnormalities |
| Skin | 55% | Hyper-hypopigmentation, café-au-lait spots |
| Growth | 60% | Short stature, endocrine disorders, IUGR. |
| Upper limbs | 43% | Absent or hypoplastic thumb or radii, hypoplastic thenar eminence, triphalangeal, absent first metacarpal, dysplastic ulnae |
| Other skeletal disorders | 43% | Microcephaly, micrognathia, sprengel, Klippel-Fiel, spina bifida, scoliosis, abnormal ribs |
| Eyes | 23% | Small, close-set, strabismus, epicanthal folds, cataracts, astigmatism |
| Renal | 21% | Ectopic or pelvic, abnormal, horseshoe, hypoplastic or dysplastic, absent, hydronephrosis or hydrourete |
| Ears | 9% | Abnormal shape, abnormal middle ear, deafness (usually conductive) |
| Gonads | 32% | Males: Hypogenitalia, undescended testes, hypospadias, micropenis |
| 3% | Females: Hypogenitalia, bicornuate uterus, abnormal menses | |
| Cardiopulmonary | 11% | Various structural congenital heart defects |
| Gastrointestinal | 11% | Atresia (esophagus, duodenum, jejunum) imperforate anus, tracheoesophageal fistula |
Table 1: Congenital abnormalities in FA patients.
Approximately 3/4 of FA patients develop marrow dysfunction, ranging from mild asymptomatic cytopenia to severe Aplastic Anemia (AA) within the first decade of life. Aplastic anemia usually occurs at a median age of 8-10 years. FA patients can develop neutropenia and thrombocytopenia as well as anemia, Myelodysplastic Syndrome (MDS) or AML [14,15]. The relative risk of AML as compared to the general population is much higher and the median age in reported cases is 13 years, with a range from birth to 50 years of age. There is a cumulative risk of more than 50% of marrow failure and 20% of AML by the age of 40. Absence of marrow dysfunction, however, does not rule out the diagnosis of FA [14-16].
Marrow cellularity is best evaluated by bone marrow biopsy and the bone marrow often appears dysplastic, but the relationship between clonal cytogenetic abnormalities and progression to leukemia is not always clear. The most common cytogenetic abnormalities observed in FA patients involve chromosomes 1,3,4 or 7, but there is a striking association between chromosome 3q26q29 amplifications (partial trisomies and tetrasomies) and rapid progression to MDS or AML [17].
Patients are also at high risk (30% by age 40) of developing specific solid tumors at an unusually young age, including head, neck, esophagus, liver tumors, gynecological squamous cell carcinomas of the lower genital tract and anal cancers [18-20]. Compared to the general population, the risk is approximately 50-fold higher for all solid tumors, but hundred- to thousand-fold higher for cancers of the head and neck, esophagus, liver, vulva, and cervix [18-21]. The mechanisms by which defects in the FA complex increase susceptibility to specific solid tumors are not completely elucidated. Genomic instability caused by subsequent somatic events occurring relatively early in the life of FA patients may explain the high rate of solid tumors in FA. It is also noteworthy that there are associations between oral androgens and liver tumors, as well as certain Human Papilloma Viruses (HPVs) and gynecologic, head and neck cancers [21-23]. The fact that approximately 25% of reported FA patients were diagnosed after developing cancer highlights the concern for early diagnosis of FA [18,22,23].
GENETICS OF FA
Sixteen genes associated with the FA phenotype have been identified since 1992. As table 2 shows molecular defects in the FANCA gene are the most frequent, followed by FANCC and FANCG. FANCB is a rare X-linked recessive gene, while all the others are autosomal recessive [24-29]. The highest frequency of molecular defects in the FANCA gene (80%) is found in Spanish Gypsies [30], while founder effects including mutations in FANCC exist in Ashkenazi Jews [31]. Alterations in FANCG gene are also present in high incidence in Portuguese-Brazilian, French Acadian, Korean/Japanese and South Africans [32].
| GENES | Molecular Defect (%) | Chromosome Location | Function |
| FANCA | 60%-70% | 16q24.3 | Partner of FANCG, contains nuclear localization signal |
| FANCB | 2% | Xp22.2 | Partner of FANCL, contains nuclear localization signal |
| FANCC | 15% | 9q22.3 | Partner of FANCE |
| FANCD1/BRCA2 | 3% | 13q12.3 | Supports RAD51 filament formation |
| FANCD2 | 3% | 3p26 | Monoubiquitinated and phosphorylated following DNA damage |
| FANCE | 3% | 6p21 | Partner of FANCC and FANCD2, CHK1 target, contains nuclear localization signal |
| FANCF | 2% | 11p15 |
Adaptor protein stabilizing A/G and E/C Interaction |
| FANCG/XRCC9 | 10% | 9p13 | Partner of FANCA |
| FANCI | 1% | 15q26.1 | Partner of FANCD2, monoubiquitinated and phosphorylated following DNA damage |
| FANCJ/BRIP1 | 2% | 17q22-24 | 5′-to-3′helicase |
| FANCL | 0.2% | 2p16.1 | E3 ubiquitin ligase |
| FANCM | 0.2% | 14q21.3 | Translocase |
| FANCN/PALB2 | 0.7% | 16p12.1 |
Partner of BRCA2, essential for stability and localization of BRCA2 |
| FANCO/RAD51C | 0.2% | 17q22 | Strand-transfer protein, involved in recombinational repair of damaged DNA and in meiotic recombination |
| FANCP/SLX4 | 0.2% | 16p13 | Structure specific endonuclease subunit |
| FANCQ/XPF | 0.2%
|
16p13.12 | Catalytic component of a structure-specific DNA repair endonuclease |
Genotype/Phenotype correlation
Monoallelic mutations in FANCD1/BRCA2, FANCJ/BRIP1, FANCN/PALB2, FANCO/RAD51C, FANCP/SLX4 and FANCQ/XPF are related with ovarian and breast cancer, but molecular defects in these genes are also identified in the non-FA population [33,42-46].
FA PATHWAY
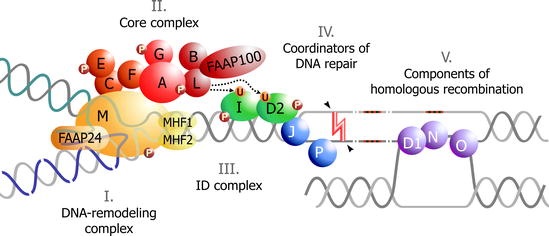
During the S phase of the cell cycle, DNA is duplicated at multiple replication forks. Interstrand Cross-Linking agents (ICL) prevent the separation of the two strands causing stalled replication forks and impairing DNA integrity [47]. The FA pathway responds to DNA damage, is involved in the process of DNA repair and the encoded proteins are grouped into 3 categories [48-51]:
- FA Core Complex (FA-CC) that includes eight FA-proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL and FANCM) and three FA associated proteins (FAAP20, FAAP24 and FAAP100).
- ID2 complex that includes the proteins FANCD2 and FANCI.
- Downstream proteins BRCA2/FANCD1, BACH1/FANCJ, PALB2/FANCN, RAD51C/FANCO, SLX4/FANCP, and XPF/FANCQ.
When responding to DNA damage FA-CC activates the monoubiquitination of FANCD2 and FANCI [51-53]. FAAP20 binds directly to FANCA and FANCG and participates in the regulation of the FA pathway [54]. FAAP24 interacts with FANCM and promotes the specific targeting of FANCM/FAAP24 to the binding sites of single-stranded DNA [55]. FAAP100 interacts with FANCB and FANCL to form a stable sub complex [56]. It is noteworthy that up to now, no mutation has been identified in FA patients in several genes encoding the FA pathway-related proteins, including FAAP20, FAAP24 and FAAP100 [54-56].
FANCL provides the E3 ubiquitin ligase activity of the FA-CC, which is necessary for the monoubiquitination of the ID complex [57]. Many of the FA-proteins are phosphorylated by the damage response kinases ATM and ATR. ATR is essential for the efficient monoubiquitination of FANCD2 and FANCI, implying an important role of ATR in the FA pathway activation [58,59].
The ubiquitinated ID complex is translocated to the chromatin at DNA damage foci at both sites and the surrounding areas [59]. The ubiquitinated ID-complex recruits endonucleases, mainly XPF/FANCQ-ERCC1, to ICLs [60]. The unhooking step of ICL repair is essential for resolving the ICL but creates a DNA Double Strand Break (DSB) that requires repair. CtBP Interacting Protein (CtIP) is shown to be a novel interacting partner of FANCD2 likely to promote DNA end resection and thus causing activation of HR or translesion synthesis, a repair mechanism that can bypass the ICL by creating a double stranded DNA molecule [61-63]. Downstream proteins have an important role in this repair mechanism, connecting the FA-pathway with DSB repair [48,49].
The remaining adducts are removed by Nucleotide Excision Repair (NER). The ERCC4 (FANCQ) gene encodes for a DNA repair endonuclease (XPF) that plays an essential role in NER and Interstrand Cross-Link Repair (ICLR) [60].
The FA-BRCA2 pathway is finally inactivated by the de-ubiquitination of the ID complex which is catalyzed by the USP1-UAF1 De-Ubiquitinase (DUB) (Figure 1) [64].
DIAGNOSIS OF FA
Early identification of FA allows differential diagnosis from other disorders, precludes inappropriate management of hematologic disease (such as AA, MDS, AML) and permits appropriate consideration of possible treatment or supportive care [4,7].
CYTOGENETIC ANALYSIS
The first diagnostic test used is the chromosomal breakage test. Two different sets of peripheral blood lymphocyte cultures - patient’s and control’s matched for age and sex- are set up and Mitomycin C (MMC) or Diepoxybutane (DEB) are added for clastogen-induced chromosome damage. As FA positive is considered the case in which the percentage of aberrations (breaks, gaps, rearrangements, radials, exchanges, and endoreduplications) in the patient’s lymphocytes is 7-10 times higher as compared to control (Figure 2) [5].

Molecular analysis
Flow cytometry
FANCD2 western blots
Complementation analysis
Next Generation Sequencing (NGS)
Multiplexed next-generation sequencing, based on massively parallel sequencing, is an effective molecular diagnostic approach for FA. The procedure, performed on genomic DNA, reduces the turnaround time, number of assays, and cost for a reliable detection of the disease-causing mutation. To increase the efficiency of the molecular diagnosis, genes involved in other bone marrow failure syndromes (e.g., Diamond-Blackfan anemia and Shwachman-Diamond syndrome) can be included, in order to diagnose these non-FA patients that are often referred for FA diagnosis [69].
MOSAICISM IN FANCONI ANEMIA
Approximately 25% of patients with FA show spontaneously occurring mosaicism with two subpopulations of lymphocytes, one of which is hypersensitive to cross-linking agents (MMC, DEB), while the other responds normally. Mosaicism, that may improve survival for the patients, can occur by mechanisms such as back mutation, intragenic crossover, gene conversion and compensating deletions/insertions. If lymphocyte analysis is inconclusive because of revertant mosaicism, fibroblasts are obtained since they do not show genetic reversion in vivo [70,71].
PRENATAL DIAGNOSIS AND GENETIC COUNSELING
Indications for prenatal testing include targeted prenatal testing when the molecular defect is known, ultrasonographic findings, or families with a prior affected child when no information is available on the molecular defects.
Chromosome breakage test
Molecular testing
Fetal ultrasound evaluation
Preimplantation Genetic Diagnosis (PGD)
Genetic counseling: Genetic counseling of families with FA depends on the FA subtype and should include family and pregnancy histories, clarify the mode of inheritance and explain the genetic testing process [78]. It must provide information about current research opportunities, support groups and future reproductive options. Family members must also be informed about the possibility of developing ovarian and breast cancer if they are carriers of molecular defects that are located in the high risk FANC genes [79]. In case of novel molecular defects, further investigation is required in order to elucidate the consequences in DNA expression, since new approaches for treatment of FA patients may arise [78].
TREATMENT
There are short-term and long-term protocols for treatment of FA patients depending on the symptoms. Since the main disorder is the progressive dysfunction of bone marrow, treatment includes:
Androgens: Approximately half of FA patients respond well to androgens, which stimulate the production of red blood cells, platelets and sometimes white cell production. It is essential that use of androgens is considered in the context of an eventual bone marrow transplant, as their use may adversely affect the ultimate success of a transplant [80].
Growth factors: Growth factors (G-CSF) stimulate the production of white blood cells [80].
Bone marrow transplantation: This is the only long-term cure for the blood defects in FA and FA was the first disease for which cord blood transplantation was introduced [81].This treatment has many risks for FA patients because of their extreme sensitivity to radiation and chemotherapy [80,82].
NEW INSIGHTS
FA presents great heterogeneity including serious congenital and hematological abnormalities and researchers worldwide try to develop diagnostic tests, effective treatments and possibly a cure for this disease. New insights for diagnosis of FA are:
Fanconi anemia antibody project: The Fanconi Anemia Research Fund, through a partnership with Oregon Health & Science University, has established the Fanconi Anemia Antibody Project, which concerns the development of antisera against the FA complementation group proteins. The technique mainly used in this research protocol is western blot [83].
MicroRNAs: MicroRNAs (miRNAs) regulate gene expression post-transcriptionally, are involved in biological processes, such as cell proliferation, differentiation and apoptosis and are deregulated in cancer. The hypothesis that microRNAs regulate the FA-BRCA pathway and promote chemosensitivity and genomic instability in cancer cells has been proven; several microRNAs inhibit Ionizing Radiation (IR)-induced nuclear foci formation of FANCD2 and/or RAD51. Specific miRNAs are deregulated and play a role in the hematopoietic dysfunction of FA patients [84]. One of those, hsa-miR-181c, interacts with the 3 UTR of TNF, inhibiting its expression and toxic effects in hematopoietic progenitors from FA patients [85]. Studies of microRNAs may lead to the discovery of novel factors that are potential targets for chemo sensitization. These may be novel Fanconi anemia genes or breast/ovarian cancer susceptibility genes that are involved in the pathway and microRNA analysis may constitute a new diagnostic tool for Fanconi Anemia.
Aldehyde blocking agents: Endogenous aldehydes are an important source of genotoxicity in the human hematopoietic system, and the FA pathway counteracts them [86]. If the FA pathway is compromised, Hematopoietic Stem Cells (HSCs) accumulate aldehyde-induced DNA damage, resulting in BMF. Some modifier genes or environmental factors might affect levels of aldehydes or other genotoxic substances. This may be the reason why some ALDH2-proficient FA patients develop BMF early [86]. ALDH2 encodes an aldehyde dehydrogenase that participates in the oxidative pathway of alcohol metabolism. Administration of ALDH2 agonists, such as Alda-1 or a similar drug could be beneficial for ALDH2-proficient FA cases, stimulating the enzymatic activity of both the normal and variant ALDH2 [87].
Over the last two decades enormous efforts in the clinical and genetic field have changed the character of FA from a life-limiting disease to a chronic condition of highly variable severity. Methods of in vitro gene therapy might complement these approaches, mainly for FA patients with molecular defects in the commonly mutated FANC genes [88,89]. Gene therapy in this form will principally address hematological problems associated with FA [90]. In addition, patient and family support groups have an essential role in this context.
REFERENCES
- Fanconi G (1927) Familiäre infantile perniziosaartige Anämie (perniziöses Blutbild und Konstitution). Jb Kinderheilk, Karger, Germany.
- Schroeder TM (1982) Genetically determined chromosome instability syndromes. Cytogenet Cell Genet 33: 119-132.
- Tomaszewska A, Srebniak M, Gny? A (2006) [Chromosome instability syndromes]. Pol Merkur Lekarski 20: 577-581.
- Giampietro PF, Adler-Brecher B, Verlander PC, Pavlakis SG, Davis JG, et al. (1993) The need for more accurate and timely diagnosis in Fanconi anemia: a report from the International Fanconi Anemia Registry. Pediatrics 91: 1116-1120.
- Oostra AB, Nieuwint AW, Joenje H, de Winter JP (2012) Diagnosis of fanconi anemia: chromosomal breakage analysis. Anemia 2012: 238731.
- Alter BP (2007) Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program 29-39.
- Auerbach AD (2009) Fanconi anemia and its diagnosis. Mutat Res 668: 4-10.
- Shimamura A, Alter BP (2010) Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 24: 101-122.
- Parikh S, Bessler M (2012) Recent insights into inherited bone marrow failure syndromes. Curr Opin Pediatr 24: 23-32.
- Zen PRG, de Moraes FN, Rosa RFM, Graziadio C, Paskulin GA (2011) Clinical characteristics of patients with Fanconi anemia. Rev paul pediatr 29: 3.
- Bagby GC, Alter BP (2006) Fanconi anemia. Semin Hematol 43: 147-156.
- Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, et al. (2003) A 20-year perspective on the International Fanconi Anemia Registry (IFAR). Blood 101: 1249-1256.
- Soulier J (2011) Fanconi anemia. Hematology Am Soc Hematol Educ Program 2011: 492-497.
- Butturini A, Gale RP, Verlander PC, Adler-Brecher B, Gillio AP, et al. (1994) Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study. Blood 84: 1650-1655.
- Rosenberg PS, Greene MH, Alter BP (2003) Cancer incidence in persons with Fanconi anemia. Blood 101: 822-826.
- Alter BP, Caruso JP, Drachtman RA, Uchida T, Velagaleti GV, et al. (2000) Fanconi anemia: myelodysplasia as a predictor of outcome. Cancer Genet Cytogenet 117: 125-131.
- Tönnies H, Huber S, Kuhl JS, Gerlach A, Ebell W, et al. (2003) Clonal chromosomal aberrations in bone marrow cells of Fanconi anemia patients: gains of the chromosomal segment 3q26q29 as an adverse risk factor. Blood 101: 3872-3874.
- Alter BP (2003) Cancer in Fanconi anemia, 1927-2001. Cancer 97: 425-440.
- Sanghvi LD, Rao DN, Joshi S (1989) Epidemiology of head and neck cancers. Semin Surg Oncol 5: 305-309.
- van Zeeburg HJ, Snijders PJ, Wu T, Gluckman E, Soulier J, et al. (2008) Clinical and molecular characteristics of squamous cell carcinomas from Fanconi anemia patients. J Natl Cancer Inst 100: 1649-1653.
- Kutler DI, Auerbach AD, Satagopan J, Giampietro PF, Batish SD, et al. (2003) High incidence of head and neck squamous cell carcinoma in patients with Fanconi anemia. Arch Otolaryngol Head Neck Surg 129: 106-112.
- Hørding U, Daugaard S, Bock JE (1992) Human papillomavirus, Epstein-Barr virus, and cervical carcinoma in Greenland. Int J Gynecol Cancer 2: 314-317.
- Touraine RL, Bertrand Y, Foray P, Gilly J, Philippe N (1993) Hepatic tumours during androgen therapy in Fanconi anaemia. Eur J Pediatr 152: 691-693.
- Vaz F, Hanenberg H, Schuster B, Barker K, Wiek C, et al. (2010) Mutation of the RAD51C gene in a Fanconi anemia-like disorder. Nat Genet 42: 406-409.
- Shamseldin HE, Elfaki M, Alkuraya FS (2012) Exome sequencing reveals a novel Fanconi group defined by XRCC2 mutation. J Med Genet 49: 184-186.
- Bogliolo M, Schuster B, Stoepker C, Derkunt B, Su Y, et al. (2013) Mutations in ERCC4, encoding the DNA-repair endonuclease XPF, cause Fanconi anemia. Am J Hum Genet 92: 800-806.
- de Winter JP, Joenje H (2009) The genetic and molecular basis of Fanconi anemia. Mutat Res 668: 11-19.
- Kim Y, Lach FP, Desetty R, Hanenberg H, Auerbach AD, et al. (2011) Mutations of the SLX4 gene in Fanconi anemia. Nat Genet 43: 142-146.
- Huret JL, Ahmad M, Arsaban M, Bernheim A, Cigna J, et al. (2013) Atlas of genetics and cytogenetics in oncology and haematology in 2013. Nucleic Acids Res 41: 920-924.
- Callén E, Casado JA, Tischkowitz MD, Bueren JA, Creus A, et al. (2005) A common founder mutation in FANCA underlies the world’s highest prevalence of Fanconi anemia in Gypsy families from Spain. Blood 105: 1946-1949.
- Verlander PC, Kaporis A, Liu Q, Zhang Q, Seligsohn U, et al. (1995) Carrier frequency of the IVS4 + 4 A-->T mutation of the Fanconi anemia gene FAC in the Ashkenazi Jewish population. Blood 86: 4034-4038.
- Auerbach AD, Greenbaum J, Pujara K, Batish SD, Bitencourt MA, et al. (2003) Spectrum of sequence variation in the FANCG gene: an International Fanconi Anemia Registry (IFAR) study. Hum Mutat 21: 158-168.
- Mathew CG (2006) Fanconi anaemia genes and susceptibility to cancer. Oncogene 25: 5875-5884.
- Schneider M, Chandler K, Tischkowitz M, Meyer S (2014) Fanconi anaemia: genetics, molecular biology, and cancer - implications for clinical management in children and adults. Clin Genet.
- Kee Y, D'Andrea AD (2012) Molecular pathogenesis and clinical management of Fanconi anemia. J Clin Invest 122: 3799-3806.
- Faivre L, Guardiola P, Lewis C, Dokal I, Ebell W, et al. (2000) Association of complementation group and mutation type with clinical outcome in fanconi anemia. European Fanconi Anemia Research Group. Blood 96: 4064-4070.
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, et al. (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297: 606-609.
- Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, et al. (2004) Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood 103: 3226-3229.
- Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, et al. (2007) Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 39: 162-164.
- Tischkowitz M, Xia B, Sabbaghian N, Reis-Filho JS, Hamel N, et al. (2007) Analysis of PALB2/FANCN-associated breast cancer families. Proc Natl Acad Sci USA 104: 6788-6793.
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, et al. (2005) BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8: 255-265.
- Nik-Zainal S, Alexandrov LB, Wedge DC, Loo PV, Greenman CD, et al., (2012) Mutational Processes Molding the Genomes of 21 Breast Cancers. Cell 149: 979-993.
- Struewing JP, Brody LC, Erdos MR, Kase RG, Giambarresi TR, et al. (1995) Detection of eight BRCA1 mutations in 10 breast/ovarian cancer families, including 1 family with male breast cancer. Am J Hum Genet 57: 1-7.
- Mahon SM (2014) Breast cancer risk associated with CHEK2 mutations. Oncol Nurs Forum 41: 692-694.
- Shah S, Kim Y, Ostrovnaya I, Murali R, Schrader KA, et al. (2013) Assessment of SLX4 Mutations in Hereditary Breast Cancers. PLoS One 8: 66961.
- Tischkowitz M, Capanu M, Sabbaghian N, Li L, Liang X, et al. (2012) Rare germline mutations in PALB2 and breast cancer risk: a population-based study. Hum Mutat 33: 674-680.
- Sasaki MS, Tonomura A (1973) A high susceptibility of Fanconi's anemia to chromosome breakage by DNA cross-linking agents. Cancer Res 33: 1829-1836.
- Yao CJ, Du W, Zhang Q, Zhang F, Zeng F, et al. (2013) Fanconi anemia pathway--the way of DNA interstrand cross-link repair. Pharmazie 68: 5-11.
- Mirchandani KD, D'Andrea AD (2006) The Fanconi anemia/BRCA pathway: a coordinator of cross-link repair. Exp Cell Res 312: 2647-2653.
- Longerich S, Li J, Xiong Y, Sung P, Kupfer GM (2014) Stress and DNA repair biology of the Fanconi anemia pathway. Blood 124: 2812-2819.
- Kennedy RD, D'Andrea AD (2005) The Fanconi Anemia/BRCA pathway: new faces in the crowd. Genes Dev 19: 2925-2940.
- Sareen A, Chaudhury I, Adams N, Sobeck A (2012) Fanconi anemia proteins FANCD2 and FANCI exhibit different DNA damage responses during S-phase. Nucleic Acids Res 40: 8425-8439.
- Sims AE, Spiteri E, Sims RJ 3rd, Arita AG, Lach FP, et al. (2007) FANCI is a second monoubiquitinated member of the Fanconi anemia pathway. Nat Struct Mol Biol 14: 564-567.
- Leung JW, Wang Y, Fong KW, Huen MS, Li L, et al. (2012) Fanconi Anemia (FA) binding protein FAAP20 stabilizes FA complementation Group A (FANCA) and participates in interstrand cross-link repair. Proc Natl Acad Sci USA 109: 4491-4496.
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, et al. (2007) Identification of FAAP24, a Fanconi Anemia Core Complex Protein that Interacts with FANCM. Mol Cell 25: 331-343.
- Ling C, Ishiai M, Ali AM, Medhurst AL, Neveling K, et al. (2007) FAAP100 is essential for activation of the Fanconi anemia-associated DNA damage response pathway. EMBO J 26: 2104-2114.
- Cole AR, Lewis LP, Walden H (2010) The structure of the catalytic subunit FANCL of the Fanconi anemia core complex. Nat Struct Mol Biol 17: 294-298.
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, et al. (2002) Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 109: 459-472.
- Moldovan GL, D'Andrea AD (2009) How the fanconi anemia pathway guards the genome. Annu Rev Genet 43: 223-249.
- Osorio A, Bogliolo M, Fernández V, Barroso A, de la Hoya M, et al. (2013) Evaluation of Rare Variants in the New Fanconi Anemia Gene ERCC4 (FANCQ) as Familial Breast/Ovarian Cancer Susceptibility Alleles. Hum Mutat 34: 1615-1618.
- Duquette ML, Zhu Q, Taylor ER, Tsay AJ, Shi LZ, et al. (2012) CtIP is required to initiate replication-dependent interstrand crosslink repair. PLoS Genet 8: 1003050.
- Murina O, von Aesch C, Karakus U, Ferretti LP, Bolck HA, et al. (2014) FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep 7: 1030-1038.
- Unno J, Itaya A, Taoka M, Sato K, Tomida J, et al. (2014) FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Rep 7: 1039-1047.
- Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, et al. (2005) The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 17: 331-339.
- Gille JJP, Floor K, Kerkhoven L, Ameziane N, Joenje H, et al. (2012) Diagnosis of Fanconi Anemia: Mutation Analysis by Multiplex Ligation-Dependent Probe Amplification and PCR-Based Sanger Sequencing. Anemia.
- Moreira CF, Brito LC Jr, Lemos JA (2008) Flow cytometry for diepoxybutane test analysis. Genet Mol Res 7: 1353-1359.
- Chandra S, Levran O, Jurickova I, Maas C, Kapur R, et al. (2005) A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol Ther 12: 976-984.
- Strathdee CA, Gavish H, Shannon WR, Buchwald M (1992) Cloning of cDNAs for Fanconi's anaemia by functional complementation. Nature 358: 434.
- Ameziane N, Sie D, Dentro S, Ariyurek Y, Kerkhoven L, et al. (2012) Diagnosis of Fanconi Anemia: Mutation Analysis by Next-Generation Sequencing. Anemia.
- Lo Ten Foe JR, Kwee ML, Rooimans MA, Oostra AB, Veerman AJ, et al. (1997) Somatic mosaicism in Fanconi anemia: molecular basis and clinical significance. Eur J Hum Genet 5: 137-148.
- Gross M, Hanenberg H, Lobitz S, Friedl R, Herterich S, et al. (2002) Reverse mosaicism in Fanconi anemia: natural gene therapy via molecular self-correction. Cytogenet Genome Res 98: 126-135.
- Auerbach AD, Min Z, Ghosh R, Pergament E, Verlinsky Y, et al. (1986) Clastogen-induced chromosomal breakage as a marker for first trimester prenatal diagnosis of Fanconi anemia. Hum Genet 73: 86-88.
- Tamary H, Bar-Yam R, Zemach M, Dgany O, Shalmon L, et al. (2002) The molecular biology of Fanconi anemia. Isr Med Assoc J 4: 819-823.
- Alter BP, Kupfer G (2002) Fanconi Anemia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, et al. (eds.). Gene Reviews. Seattle, Washington, USA.
- Verlinsky Y, Rechitsky S, Schoolcraft W, Strom C, Kuliev A (2001) Preimplantation Diagnosis for Fanconi Anemia Combined with HLA matching. JAMA 285: 3130-3133.
- Wagner JE, Davies SM, Auerbach AD (1999) Hematopoietic stem cell transplantation in the treatment of Fanconi Anemia. In: Forman SJ, Blum KG, Thomas ED, (eds.). Hematopoietic Cell Transplantation (2nd edn.). Malden, Mass: Blackwell Science Inc, USA. Pg: 1204-1219.
- Verlinsky Y, Kuliev A (2000) An Atlas of Preimplantation Genetic Diagnosis. CRC press, New York, USA.
- Rantala J, Platten U, Lindgren G, Nilsson B, Arver B, et al. (2009) Risk perception after genetic counseling in patients with increased risk of cancer. Hered Cancer Clin Pract 7: 15.
- Berliner JL, Fay AM, Practice issues subcommittee of the National Society of Genetic Counselors’ Familial Cancer Risk Counseling Special Interest Group (2007) Risk assessment and genetic counseling for hereditary breast and ovarian cancer: Recommendations of the National Society of Genetic Counselors. J Genet Couns 16: 241-260.
- Shimamura A, Alter BP (2010) Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev 24: 101-122.
- Gluckman E, Broxmeyer HA, Auerbach AD, Friedman HS, Douglas GW, et al. (1989) Hematopoietic reconstitution in a patient with Fanconi’s anemia by means of umbilical-cord blood from an HLA-identical sibling. N Engl J Med 321: 1174-1178.
- MacMillan ML, Wagner JE (2010) Haematopoeitic cell transplantation for Fanconi anaemia - when and how? Br J Haematol 149: 14-21.
- http://www.ohsu.edu/research/fanconi-anemia/index.cfm
- Huang JW, Villegas E, Taniguchi T (2010) MicroRNA-mediated regulation of the Fanconi anemia-BRCA pathway. Cancer Research 70.
- Río P, Agirre X, Garate L, Baños R, Álvarez L, et al. (2012) Down-regulated expression of hsa-miR-181c in Fanconi anemia patients: implications in TNFα regulation and proliferation of hematopoietic progenitor cells. Blood 119: 3042-3049.
- Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ, et al. (2012) Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature 489: 571-575.
- Chen CH, Budas GR, Churchill EN, Disatnik MH, Hurley TD, et al. (2008) Activation of aldehyde dehydrogenase-2 reduces ischemic damage to the heart. Science 321: 1493-1495.
- González-Murillo A, Lozano ML, Alvarez L, Jacome A, Almarza E, et al. (2010) Development of lentiviral vectors with optimized transcriptional activity for the gene therapy of patients with Fanconi anemia. Hum Gene Ther 21: 623-630.
- Rio P, Baños R, Lombardo A, Quintana-Bustamante O, Alvarez L, et al. (2014) Targeted gene therapy and cell reprogramming in Fanconi anemia. EMBO Mol Med 6: 835-848.
- Tolar J, Becker PS, Clapp DW, Hanenberg H, de Heredia CD, et al. (2012) Gene therapy for Fanconi anemia: one step closer to the clinic. Hum Gene Ther 23: 141-144.
Citation: Selenti N, Kattamis A, Kanavakis E, Kitsiou S, Mavrou A (2015) Genetic Diagnosis of Fanconi Anemia. J Hematol Blood Transfus Disord 2: 002.
Copyright: © 2015 Selenti N, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.