Hemolytic Disorders of the Newborn, Current Methods of Diagnosis and Treatment: A Review Study
*Corresponding Author(s):
Esan AJDepartment Of Haematology And Blood Transfusion Science, Federal Teaching Hospital, PMB 201, Ido-Ekiti, Nigeria
Tel:+234 8035477756,
Email:ayodelejacob4u@gmail.com
Abstract
Hemolytic disease of the newborn is an alloimmune condition that develops in a fetus, when the IgG molecules produced by the mother pass through the placenta. The underlying cause of hemolytic disease of the newborn is incompatibility between the maternal and fetal blood. This incompatibility results in the formation of antibodies in the maternal blood, which attacks the fetal Red Blood Cells (RBCs). During pregnancy, maternal antibodies are transported across the placenta and enter the fetal circulation and attack the red blood cells in the fetal circulation; the red cells are broken down and the fetus can develop reticulocytosis, hyperbilirubinemia and anemia. This fetal disease ranges from mild to very severe and fetal death from heart failure (hydrops fetalis) can occur. When the disease is moderate or severe, many erythroblasts are present in the fetal blood and so these forms of the disease can be called erythroblastosis fetalis. Hemolytic disease of the newborn can also due to non-immune conditions such as red blood cells membrane disorders like hereditary spherocytosis, hereditary elliptocytosis; red blood cells enzymes defects like Glucose-6-Phosphate Dehydrogenase deficiency (G6PD deficiency) and haemoglobinopathies like thalassemia, sickle cell. The diagnosis, acute management, and follow-up of neonates with hemolytic disease of the newborn still represent an important area of activity for maternity/neonatal services.
Keywords
INTRODUCTION
Historical overview
Mortality/Morbidity
Clinical presentation
CLASSIFICATIONS OF HEMOLYTIC DISEASE OF THE NEWBORN
Iso-Immune haemolysis
Reasons why ABO hemolytic disease of the newborn is generally less severe than Rhesus Hemolytic Disease of the Newborn HDN when ABO antibodies are consider to be more dangerous than rhesus antibodies;
- Severe ABO hemolytic disease of the newborn is much less frequent than severe rhesus hemolytic disease of the newborn, partially because ABH antigens are widely distributed throughout the body whereas the Rhesus antigens are found only on the red cells, hence, a smaller proportion of ABO antibodies is available to selectively destroy the baby’s red blood cells whereas the rhesus antibodies are concentrated on the red blood cell
- IgG anti-A and anti-B usually is IgG2 which is biologically inactive whereas rhesus antibodies involved in hemolytic disease of the newborn usually are IgG1 and IgG3
- Red blood cells sensitized by antibodies to the ABO-system are removed from the circulation by the liver, because liver is not immunologically competent and the baby’s liver is not fully developed, less damage is done to the red blood cells than in rhesus hemolytic disease of the newborn [20].
- Antibodies to the ABO-system bind complement because complement is developed in the liver and the baby’s liver is not mature, less complement is available to sensitized the baby’s red blood cells however, red blood cells that are sensitized by rhesus antigens are removed by the mother’s spleen which is highly immunogenic, as a result, the mother produces antibodies frequently with higher titer
- ABH antigens are farther from the red blood cells membrane than rhesus antigens; hence, the sensitized cells are returned to the baby’s circulation as spherocytes instead of being lysed by complement as would occur in rhesus HDN
- ABH antigens are not fully developed at birth and give weaker agglutination whereas rhesus antigens are well developed before birth [21]
Other antigens that causes hemolytic disease of the newborn: The antigens are Kell, Kidd, Duffy, MNS, C, E, c, e, Ge, Cw unexpected immune antibodies other than anti-D-k, K, Fy, S, etc. Uncommon, occurs in ~0.8% of pregnant women [25,26]. Immune alloantibodies usually due to anti-E, -c, -Kell, -Kidd or -Duffy.
Anti-K is the second most common form of severe HDN disease ranges from mild to severe over half of the cases are caused by multiple blood transfusions Anti-M very rare [27].
Non-Immune mediated hemohysis
Hereditary spherocytosis: This is the most common red blood cell membrane defect. It occurs in 1 in 5000 live births to parents of Northern European extraction, but is less often seen in other ethnic groups. The usual presentation of hereditary spherocytosis in the neonate is with unconjugated hyperbilirubinaemia. Most affected neonates are not anaemic, but a small number develop anaemia severe enough to require transfusion. Hereditary spherocytosis is inherited as an autosomal dominant trait, but around 25% of cases occur as the result of new mutations. The blood film shows moderate numbers of spherocytes with an appearance identical with that of ABO hemolytic disease of the newborn. However, disorders are distinguishable by the negative direct coomb test in hereditary spherocytosis. Whereas some babies will require one or two transfusions during the first 1-2 months of life, very few remain transfusion-dependent after this time. In the on-going assessment of such neonates, it is important to pause “top-up” of a red blood cell transfusion programme to evaluate if steady-state clinical and Hemoglobin (Hb) concentrations have been reached. All babies with hereditary spherocytosis should receive folic acid supplementation in the first year of life [28].
Hereditary elliptocytosis: This is a more complex disorder, the autosomal dominant form of hereditary elliptocytosis, (the heterozygotes) have no clinical manifestations (no anaemia and no jaundice) apart from elliptocytes on the blood film. No treatment is required and folic acid supplementation is not necessary. However, neonates who are homozygous or compound heterozygote for hereditary elliptocytosis mutations have severe hemolytic anaemia; the most important form is Hereditary Pyropoikilocytosis (HPP) [28].
Hereditary pyropoikilocytosis: Neonates with hereditary pyropoikilocytosis have more than one mutation in a red blood cell membrane protein (they may be homozygous or compound heterozygotes). Although hereditary pyropoikilocytosis is not common, it is clinically important, as it leads to severe, transfusion-dependent, haemolytic anaemia in affected neonates and infants. The diagnosis of hereditary pyropoikilocytosis can be made definitively by examining blood films from the neonate that show a high proportion of bizarre fragmented red blood cells and microspherocytes. A further useful diagnostic clue is the low mean corpuscular volume at birth (MCV <60 fl). Parental blood films normally show that one or both parents have red blood cell elliptocytosis. Red blood cell transfusion is usually necessary until the child is old enough to undergo splenectomy, to which there is an excellent response [28].
Red blood cell enzyme defects: The main red blood cell enzyme defects that present in the neonatal period are Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency and pyruvate kinase deficiency. They usually present with early-onset unconjugated hyperbilirubinaemia and can be distinguished from red blood cell membrane disorders in that there are usually no diagnostic changes on the blood film and by their inheritance pattern (G6PD deficiency being X-linked so rare in female neonates) [29].
Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency: Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency is seen in all ethnic groups but has a high prevalence in people from central Africa (20%) and the Mediterranean (10%). In neonatal Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency, jaundice usually presents within the first few days of life and is often severe. In contrast, anaemia is extremely rare and the blood film is completely normal. So the diagnosis must be made by assaying Glucose-6-Phosphate Dehydrogenase (G6PD) on a peripheral blood sample. Severe hyperbilirubinaemia is common, close monitoring of the bilirubin concentration is essential, particularly when interactions with other risk factors for neonatal hyperbilirubinaemia are present, such as hereditary spherocytosis or Gilbert’s syndrome, as kernicterus has been reported in this setting [29]. Counseling for parents of neonates with Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency about which medicines, chemicals and foods may precipitate hemolysis is also an essential follow-up activity in such families. Certain uncommon variants of Glucose-6-Phosphate Dehydrogenase (G6PD) deficiency are associated with chronic hemolysis, and for these children folic acid supplements should be given [30].
Pyruvate kinase deficiency: Pyruvate kinase deficiency is the second most common red blood cell enzymopathy in neonates. It is autosomal recessive and clinically heterogeneous, with presentation varying from hydrops fetalis, through severe early-onset neonatal hyperbilirubinaemia requiring exchange transfusion, to a mild unconjugated hyperbilirubinaemia mimicking physiological jaundice. The diagnosis is made by measuring pre-transfusion red blood cell pyruvate kinase activity. In mild cases, the pyruvate kinase activity may be relatively modestly reduced; making the diagnosis difficult and it is often useful to assay levels in the parents for confirmation. The blood film is sometimes distinctive, but more often shows non-specific changes of non-spherocytic hemolysis, and therefore it is good practice to assay pyruvate kinase in all babies with unexplained hemolysis after the common causes have been excluded. After the neonatal period, some, but not all, children are transfusion-dependent, and folic acid supplements should be given to prevent deficiency due to chronic hemolysis [31].
Neonatal hemolysis due to haemoglobinopathies: The haemoglobinopathies, except for α-thalassaemia major, do not usually present in the neonatal period. Occasional non-thalassaemic, structural α-globin and γ-globin gene mutations, which are clinically completely silent in adults and children, cause transient hemolytic anaemia in the neonate. Symptoms and signs of the major β-globin haemoglobinopathies (sickle cell disease and β-thalassaemia major) are rare in neonates, although modern techniques (High-Performance Liquid Chromatography (HPLC), isoelectric focusing) allow the diagnosis to be made on neonatal blood samples where family studies indicate that both parents are carriers.
α-Thalassaemia major: α-Thalassaemia major occurs when all four α-globin genes on chromosome 16 are deleted. It predominantly affects families of south east Asian origin and presents with mid-trimester fetal anaemia or hydrops fetalis, which is fatal within hours of delivery (occasionally babies have lived a few days). It can occasionally cause diagnostic confusion if a fetus developing the marked anaemia and hydrops characteristic of the condition is delivered preterm and then rapidly develops hyperbilirubinaemia [32,33]. Therefore, the diagnosis of α-thalassaemia major should be suspected in any case of severe fetal anaemia presenting in the second trimester and any case of hydrops fetalis with severe anaemia in which the parents come from South East Asia (it is also seen occasionally in families who originate from India, the Middle East or the Mediterranean). Checking the blood counts of the parents will immediately identify whether they are at risk of having a child with α-thalassaemia major: both parents will be carriers of a chromosome 16 in which both of the two α-globin genes are deleted and so they will have hypochromic, microcytic RBC indices (Mean Corpuscular Volume (MCV) usually <74 fl and Mean Corpuscular Hemoglobin (MCH) usually <24 pg). The diagnosis of α-thalassaemia major is confirmed by haemoglobin electrophoresis or High-Performance Liquid Chromatography (HPLC) (which shows only Hb Barts and Hb Portland; HbF and HbA are absent). The blood film shows hypochromic, microcytic red blood cells with vast numbers of circulating nucleated red blood cells. Neonatal management of α-thalassaemia major has no impact on survival unless the baby has received a planned programme of intrauterine transfusions. For fetuses/neonates treated in this way, postnatal management is the same as for β-thalassaemia-that is, life-long red blood cell transfusions or bone marrow transplantation after the age of 2 years [32,33].
α-Globin and γ-globin chain structural abnormalities: Most α-globin and γ-globin gene variants are clinically silent. Occasional α-globin gene variants may cause haemolytic anaemia in the newborn because, when the abnormal α-globin associates with γ-globin, the resultant haemoglobin is unstable, whereas, when the variant α-globin associates with β-globin, the resultant hemoglobin is stable. An example of this is hemoglobin Hasharon: α214Asp->His-γ2 is unstable, but, as γ-globin chain production is physiologically switched off and β-globin chain production predominates, the α214Asp->His-β2 produced is stable and the haemolytic anaemia completely resolves. A similar principle occurs in the γ-globin variant Hemoglobin F (HbF)-Poole which causes neonatal hemolytic anaemia, which resolves as the switch from γ-globin to β-globin occurs. These variants can be identified by hemoglobin High-Performance Liquid Chromatography (HPLC) and are worth considering when more common causes of unexplained hemolysis have been excluded [32,33].
GENETICS OF ISO-IMMUNE HAEMOLYSIS IN HEMOLYTIC DISEASE OF THE NEWBORN
Although the Rh antibody was and still is the most common cause of severe hemolytic disease of the newborn, other alloimmune antibodies belonging to Kell (K and k), Duffy (Fya), Kidd (Jka and Jkb), and MNSs (M, N, S, and s) systems do cause severe hemolytic disease of the newborn [34]. The Rh blood group system uses Fisher-Race nomenclature, and the Rh gene complex consists of three genetic loci each with two major alleles. They code for five major antigens denoted by letters, C, c, E, e, and D. Rh blood group antigens are inherited as determined by at least two homologous but distinct membrane-associated proteins. Two separate genes (RhCE and RhD), located on the short arm of chromosome 1, encode Rh proteins. Each gene is 10 exons in length, and a 96% homology between these genes is observed. Production of two distinct proteins from the RHCE gene is due to alternative splicing of messenger RNA. Rh gene complex is described by three loci, and, therefore, eight gene complexes are possible. These complexes are as follows (listed in decreasing order of frequency among whites): CDe, cde, CDE, cDe, Cde, cdE, CDE, and CdE. Expression is limited to RBCs, with an increasing density during their maturation, unlike the ABH system, which exists in a wide variety of tissues. Rh antigen is not expressed on RBC progenitors. Individuals who are Rh positive, 45% are homozygous (CDe/CDe), and 55% are heterozygous (CDe/cde) for the RhD gene. The Rh-negative phenotype represents absence of D protein on RBCs and most commonly results from deletion of the RHD gene on both chromosomes. However, the RHD gene has significant heterogeneity, and several inherited mutations and rearrangements in its structure can result in a lack of expressions of the RhD phenotype as well. Important examples of such mutations include the RHD pseudogene and RHD-CE-D hybrid gene. The former leads to a stop codon in RHD gene and results in a lack of transcription product despite all intact exons. It is found in 70% of South African blacks and in 25% of African Americans. The RHD-CE-D (Ccde) gene is also found in 22% of D-negative African Americans. It also results in an Rh positive genotype but a negative phenotype. Most Caucasians who are RhD negative have a complete deletion of RHD gene whereas only 18% of African blacks and 54% of African Americans who are RhD negative have complete deletion of the gene; the rest have above nonfunctional variants of the RHD gene [35]. Beyond the five major antigens, more than 100 antigenic variants of Rh group system have been identified. Individuals with these weak-D phenotypes comprise of 2 populations: first group (90%) that expresses normal but reduced quantities of D antigen on the RBC surface and most cannot be sensitized to produce anti-D. However, the second group (remaining 10%) known as partial-D (e.g., Cw, Du) that express partial D epitopes on RBC surface and can make anti-D and rarely experience fatal hemolytic disease of the newborn. The partial D phenotype results from amino acid sunstitution in the active RhD epitope [36]. Most women with partial-D phenotype are classified as Rh negative on routine testing and are candidates for Rh Immune Globulin (RhIG). Currently, testing of all Rh-negative women for weak expression of D is not recommended. However, Rh-negative infants born to Rh-negative women should undergo testing to detect the partial-D phenotype so that RhIG can be administered in the event of weak expression. Frequency of Rh negativity is higher in whites (15%) than in blacks (5%) and Hispanics (8%) and is rare in Eskimos, Native Americans, Japanese, and Asians, especially in Chinese individuals. The paternal heterozygosity determines the likelihood of an Rh-positive child being born to an Rh-negative mother.
Pathophysiology of iso-immune haemolysis in hemolytic disease of the newborn
Procedures such as amniocentesis, chorionic villus sampling, and cordocentesis also increase the risk of alloimmunization. Because the transplacental hemorrhage is less than 0.1 ml in most pregnancies, most women are sensitized as a result of small, undetectable fetomaternal hemorrhage. After the initial exposure to a foreign antigen, B-lymphocyte clones that recognize the RBC antigen are established. The maternal immune system initially produces antibodies of the Immunoglobulin M (IgM) isotype that do not cross the placenta and later produces antibodies of the IgG isotype that traverse the placental barrier. Predominant antibody subclass appears to be IgG1 in one third of individuals whereas a combination of IgG1 and IgG3 subclasses are found in the remaining individuals. IgG3 is more efficient in binding to reticuloendothelial cells and causing hemolysis because of its longer hinge region. This is termed the primary response and is dose dependent (documented in 15% of pregnancies with 1 ml of Rh-positive cells in an Rh-negative individual compared with 70% of pregnancies after 250 ml). A repeat exposure to the same antigen rapidly induces the production of IgG. This secondary immune response can be induced with as little as 0.03 ml of Rh-positive RBCs. The risk of Rh immunization after the delivery of the first child to a nulliparous Rh-negative mother is 16% if the Rh-positive fetus is ABO compatible with its mother, 2% if the fetus is ABO incompatible, and 2-5% after an abortion. The ABO-incompatible RBCs are rapidly destroyed in the maternal circulation, reducing the likelihood of exposure to the immune system. The degree of Rh sensitization of the mother is directly related to the amount of fetomaternal hemorrhage (i.e., 3% with 0.1 ml).
After sensitization, maternal anti-D antibodies cross the placenta into fetal circulation and attach to Rh antigen on fetal RBCs, which form rosettes on macrophages in the reticuloendothelial system, especially in the spleen. These antibody-coated RBCs are lysed by lysosomal enzymes released by macrophages and natural killer lymphocytes and are independent of the activation of the complement system. Reticulocytosis is noted when fetal Hb deficit exceeds 2 gm/dl compared with gestational age norms. Tissue hypoxia develops as fetal anemia becomes severe. When the Hemoglobin (Hb) level drops below 8 g/dL, a rise in umbilical arterial lactate occurs. When the Hb level drops below 4 g/dL, increased venous lactate is noted. Hydrops fetalis occurs when fetal Hb deficit exceeds 7 g/dL and starts as fetal ascites and evolves into pleural effusions and generalized edema. The various mechanisms responsible for hydrops are hypoalbuminemia secondary to depressed liver function, increased capillary permeability, iron overload secondary to hemolysis, and increased venous pressures due to poor cardiac function [37]. Prolonged hemolysis leads to severe anemia, which stimulates fetal erythropoiesis in the liver, spleen, bone marrow, and extramedullary sites, such as the skin and placenta. In severe cases, this can lead to displacement and destruction of hepatic parenchyma by erythroid cells, resulting in dysfunction and hypoproteinemia. Destruction of RBCs releases heme that is converted to unconjugated bilirubin. Hyperbilirubinemia becomes apparent only in the delivered newborn because the placenta effectively metabolizes bilirubin. Hemolytic disease of the newborn due to Kell sensitization results in hemolysis and suppression of erythropoiesis because the Kell antigen is expressed on the surface of erythroid progenitors. This leads to severe fetal disease at a lower maternal antibody titer than in Rhesus disease. Hemolysis associated with ABO incompatibility exclusively occurs in type-O mothers with fetuses who have type A or type B blood, although it has rarely been documented in type-A mothers with type-B infants with a high titer of anti-B IgG. In mothers with type A or type B, naturally occurring antibodies are of the IgM class and do not cross the placenta, whereas 1% of type-O mothers have a high titer of the antibodies of IgG class against both A and B. They cross the placenta and cause hemolysis in fetus. Hemolysis due to anti-A is more common than hemolysis due to anti-B, and affected neonates usually have positive direct Coombs test results. However, hemolysis due to anti-B IgG can be severe and can lead to exchange transfusion. Because A and B antigens are widely expressed in various tissues besides RBCs, only a small portion of antibodies crossing the placenta are available to bind to fetal RBCs. Recent analysis of IgG subclass in ABO incompatible direct coombs positive neonates showed IgG2 was predominent antibody which is poorly transferred across placenta and less efficient in causing hemolysis while IgG1 was noted in 22% of neonates and as a group had similar rate of hemolysis and severity of hyperbilirubinemia [38]. In addition, fetal RBCs appear to have less surface expression of A or B antigen, resulting in few reactive sites; hence the low incidence of significant hemolysis in affected neonates. This results in hyperbilirubinemia as a predominant manifestation of incompatibility (rather than anemia), and peripheral blood film frequently reveals a large number of spherocytes and few erythroblasts, unlike what is seen in Rh incompatibility (erythroblastosis fetalis), in which blood film reveals a large number of nucleated RBCs and few spherocytes [39].
Pathogenesis of hemolytic disease of the newborn: In hemolytic disease, fetal red blood cells are coated with maternal antibody. These cells are removed from the baby’s circulation by the baby’s reticuloendothelial system and destroyed. The result of hemoglobin catabolism is the production of indirect bilirubin, therefore, an increased amount of indirect bilirubin is produced, which crosses the placental and is removed by the mother’s liver. Anaemia that results from the destruction of red blood cells causes the baby’s hematopoietic tissue to produce more red blood cells and to release them prematurely. This results in an increased number of reticulocytes and nucleated red blood cells, an enlarged spleen and liver [21].
Hydrops fetalis: Hydrops fetalis is a generalized edema that affects all tissues including lungs. This condition is fatal before or after delivery. When the anaemia suffered by the fetus is severe, it causes anoxia, which causes the heart to pump harder until it fails. The heart failure is the evidence by an increase in blood volume, venous pressure, sodium chloride retention and edema. Anoxia is increased by inflammation, histamines (the release of which can be caused by complement factors C3a and C5a); and bile salts. Increase in anoxia causes increase in endothelia permeability which causes a decrease in osmotic pressure. Osmotic pressure keeps the fluids in the vessels, when the osmotic pressure decreases; it no longer counteracts the hydrostatic pressure which draws fluids into tissue [21].
Neonatal jaundice: Neonatal jaundice is jaundice that begins within the first few days after birth. Jaundice is the second greatest danger to hemolytic disease of the newborn, affected babies is from icterus gravis neonatorun (severe jaundice of the new born). These children have yellow amniotic fluid but the bilirubin is transported across the placenta to the mother’s circulation therefore the bilirubin levels are slightly reduced at birth: however, once the baby no longer has the benefit of the mother’s liver, the bilirubin begins to rise. Normally, unconjugated bilirubin is bound to albumin while being transported to the liver where it is conjugated with glucuronic acid to become conjugated or indirect bilirubin, however the baby’s gucuronosyl transferase does not start to function until approximately the third day after delivery. Consequently, the conjugated or indirect bilirubin accumulate in the baby’s system. Once the unconjugated bilirubin exceeds the ability of the albumin to bind it, it circulates as free or unbound bilirubin, unlike the water soluble direct bilirubin this unbound unconjugated bilirubin is lipid soluble and has a high affinity for central nervous system tissue. As a result of the bilirubin precipitate in the brain cells particularly in the ganglia. This condition is called kernicterus [21]. The kernicterus is often thought to occur when the indirect bilirubin is approximately 20 mg/dl or higher. Bilirubin can reach 20 to 40 mg/dl in as little as 36 hours after delivery. Although it usually takes 48 to 96 hours after delivery, if the cord blood bilirubin is 4 to 7 mg/dl, the baby is already in trouble because the bilirubin will continue to climb without the mother’s liver to conjugate it [40]. Children who are acidotic, hypoxic or premature or who have low serum albumin levels have a greater risk for unconjugated bilirubin. The symptoms of kernicterus are jaundice, lethargy, a reluctant to feed, rolling of the eyes, the high pitched cry, extending of the arm and opisthotonos (the condition in which the muscles draw the body into a bow shape). 70% of children with kerniterus develop irregular respiration a bloody froth from nose and death within 5 days. The remaining 30% may survive for months or even years, the latter the brain damage appears; the more likely the child is to live: however, death may come months later. If the child survives, brain damage may not be evident for years. This damage is permanent and irregular and manifests itself as mental retardation, deafness or possibly cerebral palsy. The severity of kerniterus is directly proportional to the bilirubin concentration. Albumin binds the unconjugated bilirubin and transports it to liver. The unbound unconjugated bilirubin increase when plasma albumin decrease or when sulphonamides or salicylates compete with the bilirubin for binding sites on the albumin. About 15 g/dl of the newborn’s albumin is available to bind bilirubin. At a pH of 7.4, approximately 22 mg/dl of bilirubin is bound. This is why kernicterus is rare when the bilirubin is less than 20 mg/dl. If the plasma direct bilirubin is greater than 3 mg/dl, a hemolytic process has taken place, if the cord blood hemoglobin is less than 10 g/dl and the cord blood bilirubin is greater than 3.5 mg/dl, the conjugating enzymes may become activated earlier than usual [21]. An increase in bilirubin that exceeds 5 mg/dl in 24 hours in a full term baby is an indication of a hemolytic process. The onset of jaundice in the first 24 hours after delivery also suggests the existence of a hemolytic process. Hemolysis is not always the result of haemolytic disease of newborn it can be caused by a number of conditions. Regardless of the cause of the hemolysis, an increase in bilirubin will ensue with hemolysis. The ability to excrete bilirubin is independent of the hemolytic process. Therefore, the intensity of the jaundice does not correlate with severity of the disease [21]. However, many babies suffer from, physiologic jaundice as a result of this immature livers. In this condition, the bilirubin generally increases during the first few days as a result of the decreased amount of glucuronosyl-transferase, which conjugates the bilirubin to form the water soluble bilirubin. The cause of normal, physiological jaundice is well understood. During life in the uterus, the red blood cells of the fetus contain a type of hemoglobin that is different than the hemoglobin that is present after birth. When an infant is born, the infant's body begins to rapidly destroy the red blood cells containing the fetal-type hemoglobin and replaces them with red blood cells containing the adult-type hemoglobin. This flood the liver with bilirubin derived from the fetal hemoglobin from the destroyed red blood cells. The liver in a newborn infant is not mature, and its ability to process and eliminate bilirubin is limited. As a result of both the influx of large amounts of bilirubin and the immaturity of the liver, bilirubin accumulates in the blood. Within two or three weeks, the destruction of red blood cells ends, the liver matures, and the bilirubin levels return to normal [21].
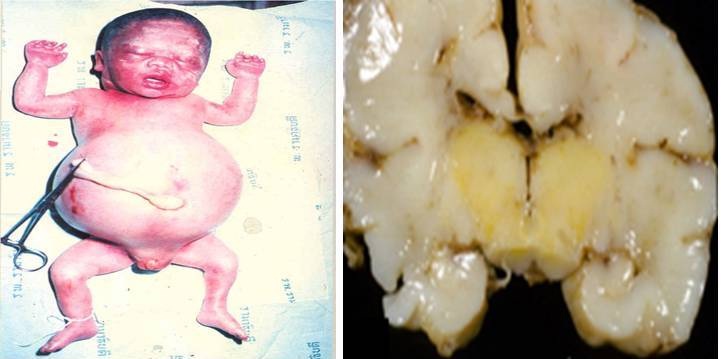
Anaemia: It is normal for an infant’s hemoglobin to decrease during first two months and even longer in premature infants. The decrease hemoglobin is presumably caused by a depression of erythropoiesis as a result of the rise in the percentage of oxygen saturation of the blood following the onset of respiration. The decrease in the hemoglobin is even more rapid in hemolytic disease of newborn, partially because of the increased red blood cell destruction and partially because erythropoiesis is suppressed as long as the hemoglobin is greater than 10 g/dl [21] (Figure 1).
RHESUS IMMUNE RESPONSE
Primary immune response: The primary immune response develops slowly. In experimental Rhesus immunization of male volunteers, 8 to 9 weeks elapsed before the response was apparent; indeed, it may not be detectable for 6 months. The primary response usually is weak and predominantly Immunoglobulin M (IgM). IgM anti-D does not cross the placenta and does not cause fetal red blood cell hemolysis. Most Rhesus-negative women convert to IgG anti-D production. IgG anti-D traverses the placenta and does produce fetal red blood cell hemolysis.
Secondary immune response: Once the primary response has occurred, a second exposure to Rhesus-positive red blood cells produces a rapid (within days) increase in anti-D, which is for the most part IgG. Subsequent exposure may produce even higher levels. If the intervals between antigen exposure are long, the subsequent exposure commonly is associated with a marked increase in Rhesus-antibody titer and increased avidity (binding ability) of the antibody for Rhesus antigen. The greater the avidity of the Rhesus antibody for Rhesus antigen (binding constant), the greater is the severity of Rhesus hemolytic disease.
Degree of severity of hemolytic disease of newborn
Degree of severity of hemolytic disease of the newborn (erythroblastosis) is determined by the amount of maternal anti-D IgG present, its avidity for the D antigen (binding constant), and the ability of the erythroblastotic fetus to replace the hemolyzed red blood cells without developing hepatic damage, portal obstruction, and hydrops fetalis.
Classification of severity of hemolytic disease of newborn (Erythroblastosis)
See Classification of severity of hemolytic disease of newborn (Erythroblastosis) in table 1.
| Degree of Severity | Incidence (%) |
| Mild: No treatment is required. Indirect bilirubin does not exceed 16-20 mg/dl. Anemia is minimal. | 45-50 |
| Moderate: Fetal hydrops will not develop. Anemia is moderate. There is severe jaundice and risk of kernicterus unless infant is treated after birth. | 25-30 |
| Severe: Fetal hydrops will develop in utero. | 20-25 |
| Before 34 weeks gestation | 10-12 |
| After 34 weeks gestation | 10-12 |
Table 1: Classification of severity of hemolytic disease of newborn (Erythroblastosis).
Mild: About half of the affected infants are mildly or not anemic at birth (cord hemoglobin exceeds 12 g/dL). They are not dangerously hyperbilirubinemic (cord bilirubin levels are less than 3.5 mg/dL). Their red blood cells, however, are coated with anti-D, making them direct antiglobulin (Coombs’ test) positive; the hallmark of all types of alloimmune hemolytic disease other than ABO [41]. In these infants, significant anemia (less than 10 g/dL) does not develop, nor does significant hyperbilirubinemia (more than 20 mg/dL in mature infants, 15 to 18 mg/dL in premature infants). They require no treatment and survive intact, as they did 60 years ago before the Rhesus blood group system was known and before any treatment was available [41].
Moderate: In about 25% of affected infants, the disease is more severe. Erythropoiesis is adequate to prevent severe anemia but not so great that hepatic hypertrophy and circulatory obstruction occur. The fetus is delivered in good condition at or near term. Before birth, the products of fetal red blood cell hemolysis are transported across the placenta and metabolized by the mother. After delivery, infants must rely on their own resources to metabolize and excrete the products of red blood cell destruction [25].
When red blood cell is destroyed, globin is split from hemoglobin, leaving the prosthetic pigment heme. Heme is rapidly converted to indirect bilirubin, which is neurotoxic. With increased red blood cell destruction, there is increased production of indirect bilirubin. At birth, an infant’s ability to metabolize indirect bilirubin is severely limited. Infants’ livers are deficient in the transport protein and the microsomal enzyme uridine diphosphoglucuronyl transferase, which are responsible for the transfer of indirect bilirubin across the liver cell membrane into the cytoplasm and then for the conjugation into water-soluble, nontoxic bilirubin diglucuronide (direct bilirubin), which is then excreted by way of the biliary canaliculi down the bile ducts into the small intestine [14].
Indirect bilirubin is water insoluble and lipid soluble and can circulate only if it is bound to the plasma protein carrier albumin. When the bilirubin-binding sites on albumin are saturated, unbound free indirect bilirubin appears. It cannot remain in a watery medium such as plasma and diffuses into high lipid content tissues. Free indirect bilirubin diffuses across the lipid-containing neuron membrane and interferes with vital metabolic processes, causing ballooning of the neural mitochondria. The neuron dies. Bilirubin accumulates within the dead neurons, producing the characteristic yellow color found at autopsy (kernicterus). Infants who develop bilirubin encephalopathy (kernicterus) are deeply jaundiced. Signs of brain dysfunction appear on the third to the fifth day. They become lethargic and hypertonic. Their necks are extended, and their wrists, elbows, and knees are flexed. They are rigid and suck poorly and they may convulse. Death occurs in 90% of such infants. In the remaining 10% of infants, jaundice lessens, and hypertonicity diminishes. Initially, the neonates appear well. As they grow older, signs of severe neurologic damage inevitably appear. Most are profoundly deaf. Most have cerebral palsy of the spastic choreoathetoid type. Intellectual retardation is variable and may be severe. Even when retardation is mild, learning and functioning are impeded by the severe deafness and abnormal movements produced by the spastic choreoathetosis [14].
Severe: The remaining 20% to 25% of erythroblastotic fetuses are so severely affected that they develop ascites and generalized edema (hydrops fetalis). Hydrops develops in half of these fetuses between 18 and 34 weeks’ gestation and in the other half between 34 and 40 weeks’ gestation.
The earlier belief that hydrops was due to hypervolemic heart failure may not be correct. Although treatment measures carried out to keep hydropic neonates alive inevitably precipitate heart failure, at birth, most hydropic infants are not hypervolemic, nor are they in heart failure. Hepatic hypertrophy with portal hypertension and hepatocellular damage is a more likely cause of hydrops fetalis [24].
With severe red blood cell destruction, hepatic erythropoiesis and hepatic enlargement become extreme. Hepatic cords and hepatic circulation are distorted by erythropoietic tissue. Portal and umbilical venous hypertension develops. The placenta becomes thick and edematous; cytotrophoblast persists. Placental perfusion is reduced, and ascites appears. As progressively greater distortion of hepatic cords by islets of erythropoiesis occurs, hepatic circulation and hepatocyte function are reduced. Hypoalbuminemia develops and produces generalized anasarca. Pleural and pericardial effusions appear. Death in utero usually occurs. If a hydropic fetus is born alive, ventilation usually is impossible because of pulmonary edema and compression hypoplasia of the lungs [24,25].
SYMPTOMS OF HEMOLYTIC DISEASE OF THE NEWBORN
During pregnancy symptoms may include
- With amniocentesis, the amniotic fluid may have a yellow coloring and contain bilirubin
- Ultrasound of the fetus shows enlarged liver, spleen, or heart and fluid buildup in the fetus’ abdomen
After birth, symptoms may include
- A pale coloring may be evident, due to anemia
- Jaundice or yellow coloring of amniotic fluid, umbilical cord, skin, and eyes may be present. The baby may not look yellow immediately after birth, but jaundice can develop quickly, usually within 24 to 36 hours
- The newborn may have an enlarged liver and spleen
- Babies with hydrops fetalis have severe edema (swelling) of the entire body and are extremely pale
- They often have difficulty breathing [4]
- Complications of hemolytic disease of the newborn (symptoms) can range from mild to severe.
During pregnancy
- Mild anemia, hyper-bilirubinemia, and jaundice: The placenta helps rid some of the bilirubin, but not all.
- Severe anemia with enlargement of the liver and spleen: When these organs and the bone marrow cannot compensate for the fast destruction of red blood cells, severe anemia results and other organs are affected.
- Hydrops fetalis: This occurs as the baby’s organs are unable to handle the anemia. The heart begins to fail and large amounts of fluid buildup in the baby’s tissues and organs. A fetus with hydrops is at great risk of being stillborn [40].
After birth
- Severe hyperbilirubinemia and jaundice: The baby’s liver is unable to handle the large amount of bilirubin that results from red blood cell breakdown. The baby’s liver is enlarged and anemia continues.
- Kernicterus: Kernicterus is the most severe form of hyperbilirubinemia and results from the buildup of bilirubin in the brain. This can cause seizures, brain damage, deafness, and death [40].
Laboratory diagnosis of hemolytic disease of the newborn
Prenatal testing
- To detect the presence of any IgG antibody that could cause hemolytic disease of the newborn in order to identify, monitor, and treat the infant as soon as possible
- To determine which women are candidates for Rhesus Immune Globin (RhIg) in order to prevent the production of anti-D [43]
- Maternal antibody titer is a good indicator of severity of hemolytic disease of the newborn
- Ultrasound - to detect organ enlargement or fluid buildup in the fetus. Ultrasound is used to view internal organs as they function, and to assess blood flow through various vessels
- Amniocentesis - to measure the amount of bilirubin in the amniotic fluid
- Sampling of some of the blood from the fetal umbilical cord during pregnancy to check for antibodies, bilirubin, and anemia in the fetus [44]
Father testing
- Investigative tests on the father depend on which maternal antibodies are present. Mother has anti-D ABO and Rhesus typing with anti-D, -C, -E, -c,-e to determine probable Rhesus genotype to predict the chance the fetus has of being Rhesus positive and affected by hemolytic disease of the newborn
- Test for weak D if initial Rhesus typing appears to be D-negative. For D positive fathers, the probable Rhesus genotype can be determined using serologic tests, i.e., DCEce typing to determine if the father is probably homozygous or heterozygous for D
- Other maternal clinically significant antibodies phenotype father for the corresponding antigen and its antithetical antigen (e.g., K and k)
Molecular genotyping
- For women who type as weak D in serologic tests, to determine the Rhesus genotype of the mother to identify if she is partial D or weak D
- For women who have made anti-D, to determine the Rhesus genotype of the father to see if fetal monitoring is needed
- For women who have made anti-D, to determine the Rhesus type of the fetus if the father is heterozygous for Rhesus D or unavailable for testing [46]
- Fetal blood typing can be done using fetal DNA from cells obtained by amniocentesis or by testing cell-free, fetal-derived DNA present in maternal plasma at 5 weeks gestation and later
- Testing of the baby’s umbilical cord blood for blood group, Rhesus factor, red blood cell count, and antibodies
- ABO blood group system (anti-A and anti-B)
- Rhesus system (Rhesus D, E, e, C and antibody combinations (i.e., anti-Rhc and anti-RhE antibodies occurring together) - can be severe
- Kell system (anti-Kell , anti-K 1 antibodies - common and also anti-K 2, anti-K 3 and anti-K 4 antibodies - rare
- Other blood group antibodies (Kidd, Lewis, Duffy, MN, P and others). Test on fetal blood (or umbilical cord blood)
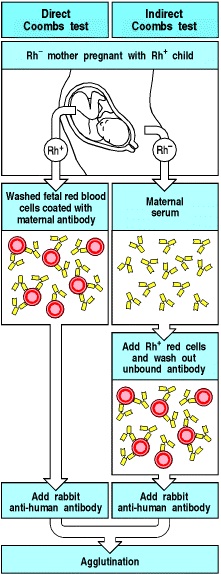
- The Direct Antiglobulin Test (DAT) is an important tool for identification of Hemolytic Disease of the Newborn (HDN) caused by erythrocyte immunization [47]. The direct Coombs test is used to confirm that the fetus or neonate has an immune mediated hemolytic anemia
- Full blood count - the hemoglobin level and platelet count are important
- Peripheral blood morphology shows increased reticulocytes. Erythroblasts (also known as nucleated red blood cells) occur in moderate and severe disease
- Biochemistry tests for jaundice: Bilirubin (total and indirect) [48]
- The Acid elution technique of Kleihauer-Betke test or flow cytometry on a postnatal maternal blood sample can confirm that fetal blood has passed into the maternal circulation and can also be used to estimate the amount of fetal blood that has passed into the maternal circulation
- The indirect Coombs test is used to screen blood from antenatal women for IgG antibodies that may pass through the placenta and cause hemolytic disease of the newborn (Figure 2)
- Rapidly progressive severe hyperbilirubinemia or prolonged hyperbilirubinemia
- Positive maternal antenatal antibody findings and/or diagnosis of anemia or fetal hydrops
- Positive neonatal direct Coombs test (direct antiglobulin test)
- Hemolysis on blood film findings [49]
- The severity of hematologic abnormalities is directly proportional to the severity of hemolysis and the extent of hematopoiesis.
- Anemia: Measurements are more accurate using central venous or arterial samples rather than capillary blood.
- Increased nucleated RBCs, reticulocytosis, polychromasia, anisocytosis, spherocytes, and cell fragmentation
- The reticulocyte count can be as high as 40% in patients without intrauterine intervention.
- The nucleated RBC count is elevated and falsely elevates the leukocyte count, reflecting a state of erythropoiesis.
- Spherocytes (<40%) are more commonly observed in cases of ABO incompatibility. Glucose does not correct the autohemolysis in ABO incompatibility unlike hereditary spherocytosis.
- In severe hemolytic disease, schistocytes and burr cells may be observed, reflecting ongoing disseminated intravascular coagulation.
- A low reticulocyte count is observed in fetuses provided with intravascular transfusion in utero and with Kell alloimmunization.
- Abnormally elevated Mean Cell Hemoglobin Concentration (MCHC) and Red Cell Distribution Width (RDW) values should prompt a diagnosis of hereditary spherocytosis [50]
- Neutropenia: This condition seems to be secondary to stimulation of erythropoiesis in favor of myelopoiesis. However, neutrophilia can be observed after intrauterine transfusion because of an increase in circulating cytokines (granulocyte-macrophage colony-stimulating factor)
- Thrombocytopenia: This condition is common, especially after intrauterine or exchange transfusions because of platelet-poor blood product and suppression of platelet production in favor of erythropoiesis
- Hypoglycemia is common and is due to islet cell hyperplasia and hyperinsulinism. The abnormality is thought to be secondary to release of metabolic byproducts such as glutathione from lysed RBCs. Hypokalemia, hyperkalemia, and hypocalcemia are commonly observed during and after exchange transfusion.
- Indirect Coombs test and direct antibody test results are positive in the mother and affected newborn. Unlike Rh alloimmunization, direct antibody test results are positive in only 20-40% of infants with ABO incompatibility. In a recent study [51], positive direct antibody test findings have a positive predictive value of only 23% and a sensitivity of only 86% in predicting significant hemolysis and need for phototherapy, unless the findings are strongly positive (4+). This is because fetal RBCs have less surface expression of type-specific antigen compared with adult cells. A prospective study has shown that the titers of maternal Immunoglobulin G (IgG) anti-A or anti-B may be more helpful in predicting severe hemolysis and hyperbilirubinemia. The sensitivity and specificity of IgG titers of 512 or higher in predicting need for invasive intervention was 90% and 73%, respectively [52]. Although the indirect Coombs test result (neonate’s serum with adult A or B RBCs) is more commonly positive in neonates with ABO incompatibility, it also has poor predictive value for hemolysis. This is because of the differences in binding of IgG subtypes to the Fc receptor of phagocytic cells and, in turn, in their ability to cause hemolysis.
- IgG2 is more commonly found in maternal serum but has weak lytic activity, which leads to the observation of little or no hemolysis with a positive direct antibody test result. On the other hand, significant hemolysis is associated with a negative direct antibody test result when IgG1 and IgG3 are predominant antibodies, which are in low concentration but have strong lytic activity, crossing to neonatal circulation.
- In newborns with hemolytic disease due to anti-c or anti-C antibodies, direct antibody test results may be negative, and the diagnosis is established after indirect Coombs testing.
TREATMENT FOR HEMOLYTIC DISEASE OF THE FETUS AND NEWBORN
Antenatal (during pregnancy), treatment for hemolytic disease of the newborn may include
Serial Ultrasound and Doppler examinations - to detect signs of fetal anemia such as increased blood flow velocities and monitor hydrops fetalis [54].
- Quantitative analysis of maternal anti-RhD antibodies - an increasing level is a sign of fetal Rhesus disease Intrauterine blood transfusion
- This is done by placing a needle through the mother’s uterus and into the abdominal cavity of the fetus or directly into the vein in the umbilical cord. Intraperitoneal transfusion - blood transfused into fetal abdomen
- Intravascular transfusion - blood transfused into fetal umbilical vein [55]
- Early delivery (usually after about 36 wks gestation) If the fetus has mature lungs, labor and delivery may be induced to prevent worsening of hemolytic disease of the newborn
- Nonstress Test (NST)
- Ultrasonographic measurement of the AFV
- Observation of the presence or absence of fetal breathing movements
- Gross body movements
- Tone [60]
| BiophysicalVariable | Normal(Score = 2) | Abnormal(Score = 0) |
| Fetal breathing movements | 1 or more episodes of ≥20s within 30 min | Absent or no episode of ≥20s within 30 min |
| Gross body movements | 2 or more discrete body/ limb movements within 30 min (episodes of active continuous movement considered as a single movement) | <2 episodes of body/limb movements within 30 min |
| Fetal tone | 1 or more episodes of active extension with return to flexion of fetal limb(s) or trunk (opening and closing of hand considered normal tone) | Slow extension with return to partial flexion, movement of limb in full extension, absent fetal movement, or partially open fetal hand |
| Reactive fhr | 2 or more episodes of acceleration of ≥15 Beats Per Minute (BPM) and of >15s associated with fetal movement within 20 min | 1 or more episodes of acceleration of fetal heart rate or acceleration of <15 bpm within 20 min |
| Qualitative afv | 1 or more pockets of fluid measuring ≥2 cm in vertical axis | Either no pockets or largest pocket <2 cm in vertical axis |
Table 2: Criteria for coding fetal biophysical variables as normal or abnormal.
| Activity | FHR Accelerations | Tone | Gross Movement | Fetal Breathing | AFV |
| Fetal sleep | ⇓ | ⇓ | ⇓ | ⇓ | |
| Early gestational age (<33 wk) | ⇓ | ⇓ | |||
| Late gestational age (>42 wk) | ⇓ | ⇓ | ⇓ | ⇓ | |
| Maternal glucose ingestion | — | — | ⇑ | ||
| Maternal alcohol ingestion | ⇓/— | ⇑/— | |||
| Maternal magnesium administration | ⇓ | ⇓ | |||
| Artificial rupture of membranes | ⇓ | ⇓ | |||
| Premature rupture of membranes | ⇓ | ||||
| Labor | ⇓ |
PHYSIOLOGIC BASIS OF BIOPHYSICAL MONITORING:
Application of the biophysical profile:
Postnatal (after birth), treatment may include
- Blood transfusions (for severe anemia)
- Intravenous fluids (for low blood pressure)
- Help for respiratory distress using oxygen or a mechanical breathing machine
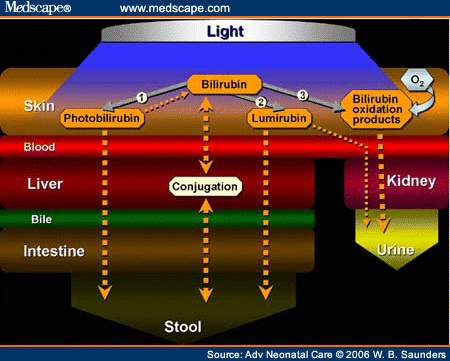
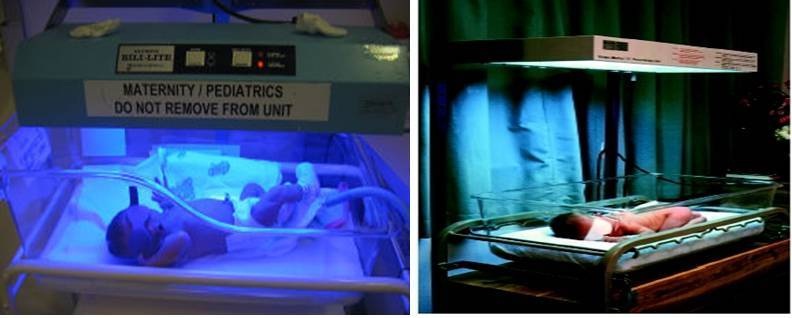
- Phototherapy: Intensive phototherapy has also been used to treat moderate and severe hemolytic disease of the newborn and decrease the need for exchange transfusion. The newborn is placed under a “blue light” which chemically alters the bilirubin in the surface capillaries to a harmless substance [63,64]
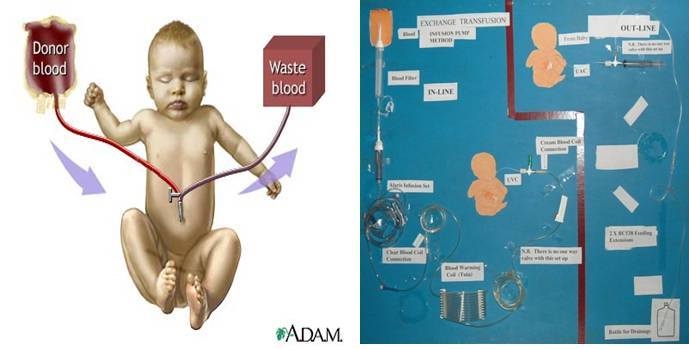
- Exchange transfusion to replace the baby’s damaged blood with fresh blood: The exchange transfusion helps increase the red blood cell count and lower the levels of bilirubin. An exchange transfusion is done by alternating giving and withdrawing blood in small amounts through a vein or artery
- Human Serum Albumin: Human serum albumin can also be transfused, either separately or as part of an exchange transfusion in place of Fresh Frozen Plasma (FFP). Albumin binds unconjugated bilirubin, thus preventing its deposition in the fat-rich brain cells. Albumin must be used judiciously, because it can aggravate congestive heart failure
Intrauterine transfusion
- Intrauterine transfusion is the only hope that babies who are severely affected before 34 weeks gestation have for survival (Figure 3)
- It provides rhesus negative cells to correct anaemia, anoxia, to prevent hydrops fetalis and fetal death.
- It can be done as early as 17 weeks, although preferable to wait until 20 weeks [55]
- Severely affected fetus, transfusions is done every 1 to 4 weeks until the fetus is mature enough to be delivered safely. Amniocentesis may be done to determine the maturity of the fetus’s lungs before delivery is scheduled
- After multiple intrauterine transfusions, most of the baby’s blood will be Rhesus D negative donor blood, therefore, the direct antiglobulin test will be negative, but the indirect antiglobulin test will be positive.
- After intrauterine transfusions, the cord bilirubin is not an accurate indicator of rate of hemolysis or of the likelihood of the need for post-natal exchange transfusion
- The mother is usually given antibiotics to prevent infection. She may also be given tocolytic medication to prevent labor from beginning, though this is unusual
- Increasingly common and relatively safe procedure since the development of high resolution ultrasound particularly with colour doppler capability (Figure 4)
- MCA Doppler velocity as a reliable non-invasive screening tool to detect fetal anemia
- The vessel can be easily visualized with color flow doppler as early as 18 weeks’ gestation
- In cases of fetal anemia, an increase in the fetal cardiac output and a decrease in blood viscosity contribute to an increased blood flow velocity [54]
- Titer greater than 32 for anti-D and 8 for anti-K OR fourfold increase in titer indicates need for analysis of amniotic fluid
- Amniocentesis
- Perform at 28 wks if HDN in previous child
- Perform at 22 wks if previous child severely affected
- Perform if maternal antibody increases before 34th wk.
- High values of bilirubin in amniotic fluid analyses by the Liley method or a hemoglobin concentration of cord blood below 10.0 g/ml
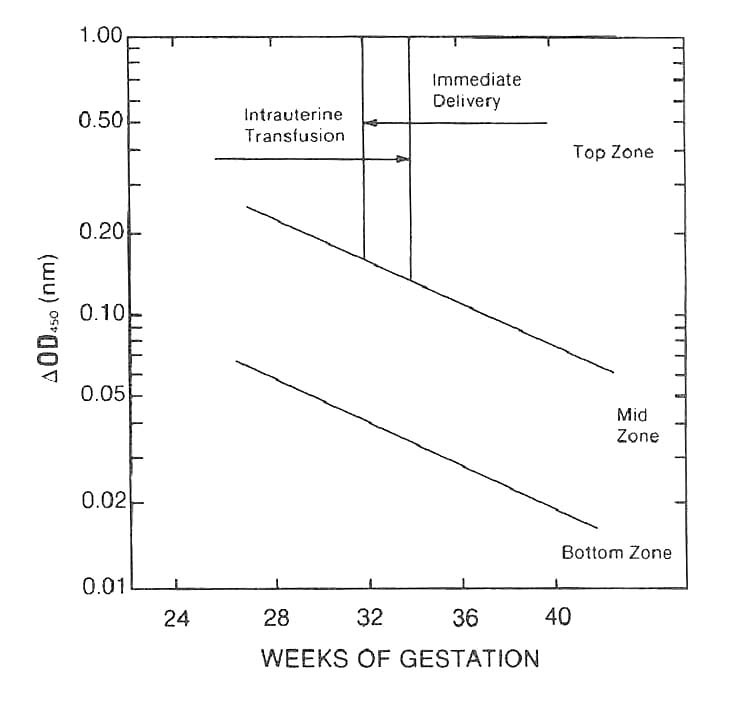
- Values remain in the bottom zone from Liley graph indicate that the baby will probably be unaffected or mild affected, values in the mid zone indicate that the baby will be moderately affected and is a candidate for early delivery and an exchange transfusion hence, values greater than 0.3 or tending from mid zone to the top zone require immediate attention (Figure 5)
- After 32 to 34 weeks gestation, delivery and exchange transfusion on the baby should be considered, before 32 weeks gestation an intrauterine transfusion is probably the best
- Type fetus -recent development in fetal RhD typing involves the isolation of free fetal DNA in maternal serum. In the United Kingdom, this technique has virtually replaced amniocentesis for fetal RhD determination in the case of a heterozygous paternal phenotype
- Maternal plasma exchange may be instituted if the fetus is too young for intrauterine transfusion
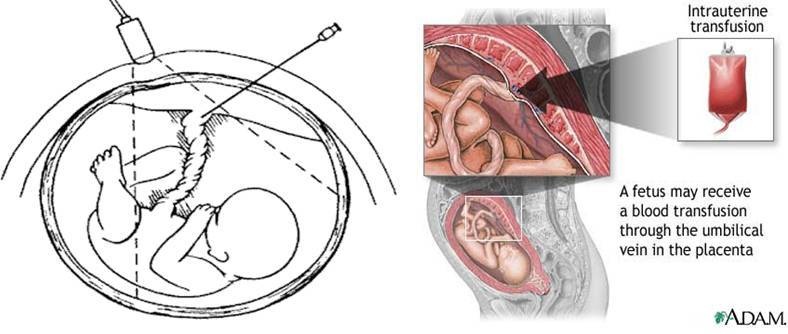
Figure 3: Intrauterine transfusion.
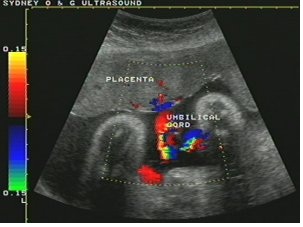
Figure 4: Ultrasound.
Exchange transfusion
- Infant weight in kilograms × 85 × Two exchanges = Total volume of blood needed
- Volume of blood needed for the exchange ÷ Two = Volume of RBC’s needed for a haematocrit of 0.50
- Volume of RBC’s needed for a haematocrit of 0.50 ÷ 0.70 = Volume of original RBC component needed with haematocrit of 0.70
- Volume of fresh frozen plasma needed = Total volume needed - Volume of RBC component needed
- The RBC-plasma combination should be mixed during the procedure to maintain the haematocrit
Haematocrit and a bilirubin should be performed on a sample the last aliquot removed to monitor the procedure. Hazards associated with exchange transfusion are numerous. The danger of air emboli and perforation of the umbilical vein are obvious, exchange also may disturb the acid-base balance, thereby causing a decrease in the pH. This is more of a problem in babies with inadequate ventilation by a high pCO2 but for most babies, a disturbed pH may be less of a problem because of the use of citrate phosphate dextrose adenine anticoagulant, on the other hand severe hypoglycemia has become more of a problem because of citrate phosphate dextrose. Sugar in anticoagulant increase the plasma glucose which causes insulin to increase and act on the glucose, causing the glucose level to drop. Heparin is not recommended because it increases the risk of bleeding and because it increases the plasma free fatty acids which increase the unconjugated bilirubin. Citrate toxicity is a potential problem that has not been substantiated. This can be diminished by infusing packed RBC’s instead of whole blood. Potassium toxicity can be avoided by using fresh blood. The uses of packed RBCs also help to prevent this problem [21].
Objective for performing exchange transfusion
- To decrease the level of bilirubin and prevent kernicterus
- To remove baby’s sensitized red blood cells
- To provide compatible rbcs with adequate oxygen carrying capacity
- To decrease the level of the incompatible antibody in the baby
Criteria for selection of donors for exchange transfusion
- Must lack RBCs antigens corresponding to the mother’s immunoglobulin G antibody
- Must lack hemolytic anti-A, anti-B and anti-AB
- Must lack unexpected antibodies active at 37°C
- Must be less than 7 day old
- Must be cross matched initially with mother’s or baby’s sermon or plasma if antibodies are present or if the donor is not group O
- Should lack hemoglobin S if the baby is acidotic or hypoxic: Testing is usually necessary only if haemoglobin S is common in the community
- Should be negative for Cytomegalovirus (CMV) if cytomegalovirus is a problem in the area and if the baby weighs less than 1200 g at birth and if the baby or mother lacks cytomegalovirus antibodies or if this information is unknown [21]
Simple transfusion
Phototherapy
Transcutaneous monitoring
- Transcutaneous bilirubinometry can be adopted as the first-line screening tool for jaundice in well, full-term babies
- This leads to about 50% decrease in blood testing (Figure 9)
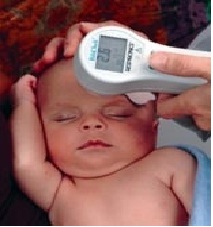
Figure 9: Transcutaneous monitoring.
Immunomodulators
Management of Hemolytic Disease of the Newborn
- Severe anaemia: Hb
- Severe hyperbilirubinaemia (total serum bilirubin >350 μmol/l) in the first 48 h of life or rapidly increasing hyperbilirubinaemia (total serum bilirubin increasing >10 μmol/l per hour)
Management of the sensitized neonate
Management of the sensitized neonate
Management of ABO incompatibility
Tin or zinc protoporphyrin or mesoporphyrins have been studied in newborns. They must be administered intramuscularly in a dose based on body weight, and their effectiveness appears to be dose related in all gestations [74]. Their possible toxic effects include skin photosensitization, iron deficiency, and possible inhibition of carbon monoxide production. Their use in Rh hemolytic disease of newborn has not been reported. Their routine use cannot be recommended yet because of lack of long-term safety data.
Furthermore, Molecular methods exist for typing Rhesus (RHD and RHCE), Kell (K & k), Duffy (Fya & Fyb), and Kidd (Jka & Jkb) loci. In prenatal testing programs, molecular typing can determine the Rhesus type of the mother, father, and fetus and may be done if the mother has anti-D or another antibody known to cause hemolytic disease of the newborn. Fetal blood typing can be done using free fetal DNA from cells obtained by amniocentesis or by testing cell-free, fetal-derived DNA present in maternal plasma at 5 weeks gestation and later [45,46].
CONCLUSION
The spectrum of hemolytic disease of the newborn has changed over the last few decades; however, the diagnosis, acute management, and follow-up of neonates with hemolytic disease of the newborn still represent an important area of activity for maternity/neonatal services. Following the introduction of prophylactic Rhesus IG in Rhesus D negative women, the incidence of Rhesus D alloimmunization in pregnancy has decreased. Consequently, hemolytic disease due to ABO incompatibility and other alloantibodies have now emerged as the major causes of hemolytic disease of the newborn. However, with increasing knowledge at the molecular level, and closer liaison between neonatal pediatricians and hematologists, the diagnosis of non-immune causes of hemolytic disease of the newborn is increasing, as these conditions have an inherited basis and therefore have implications for other family members (or future children), it must remain a high priority for all neonatal pediatricians to achieve an accurate diagnosis in all cases of hemolytic disease of the newborn and emphasizes the point that patients with persistent neonatal hemolysis should always be fully investigated and followed up appropriately.
REFERENCES
- Hendrickson JE, Delaney M (2016) Hemolytic Disease of the Fetus and Newborn: Modern Practice and Future Investigations. Transfus Med Rev.
- Kennedy MS, Krugh D (2005) Hemolytic disease of the newborn and fetus. In: Harmening DM (ed.). Modern Blood Banking and Transfusion practices, (5th edn), FA Davis, Philadelphia, USA.
- Wang M, Hays T, Ambruso DR, Silliman CC, Dickey WC (2005) Hemolytic disease of the newborn caused by a high titer anti-group B IgG from a group A mother. Pediatr Blood Cancer 45: 861-862.
- Mollison PL, Engelfret CP, Contreras M, editors (2005) Blood transfusion in clinical medicine, (11th edn), Oxford Blackwell Science, New Jersey. Pg no: 508.
- Poole J, Daniels G (2007) Blood group antibodies and their significance in transfusion medicine. Transfus Med Rev 21: 58-71.
- Thompson S, Eggington J, Dodd A (2003) Late developing red cell antibodies in pregnancy. Transfusion Medicine 13: 8-9.
- Onesimo R, Rizzo D, Ruggiero A, Valentini P (2010) Intravenous Immunoglobulin therapy for anti-E hemolytic disease in the newborn. J Matern Fetal Neonatal Med 23: 1059-1061.
- Stockman JA, de Alarcon PA (2001) Overview of the state of the art of Rh disease: history, current clinical management, and recent progress. J Pediatr Hematol Oncol. 23: 385-393.
- Bowman JM (1999) Hemolytic disease (erythroblastosis fetalis) In: Creasy RK, Resnik R (eds.). Maternal-fetal medicine (4th edn), Saunders, Philadelphia, USA. Pg no: 736-767.
- Lindenburg IT, Smits-Wintjens VE, van Klink JM, Verduin E, van Kamp IL, et al. (2012) Long-term neurodevelopmental outcome after intrauterine transfusion for hemolytic disease of the fetus/newborn: the LOTUS study. Am J Obstet Gynecol 206: 141.
- Gruslin-giroux A, Moore TR (2006) Erythroblastosis Fetalis. In: Fanaroff AA, Martin RJ (eds.). Neonatal-Perinatal Medicine, (8th edn), St. Louis, MO: Mosby-year book Inc, Maryland Heights, USA. Pg no: 389-408.
- Moise KJ (2008) Fetal anemia due to non-Rhesus-D red-cell alloimmunization. Semin Fetal Neonatal Med 13: 207-214.
- Alcock GS, Liley H (2002) Immunoglobulin infusion for isoimmune haemolytic jaundice in neonates. Cochrane Database Syst Rev.
- Basu S, Kaur R, Kaur G, Jain S (2011) Severe hemolytic disease of newborn due to non Rh D antibody. Indian J Pediatr 78: 366-367.
- Jeon H, Calhoun B, Pothiawala M, Herschel M, Baron BW (2000) Significant ABO hemolytic disease of the newborn in a group B infant with a group A2 mother. Immunohematology 16: 105-108.
- Ziprin JH, Payne E, Hamidi L, Roberts I, Regan F (2005) ABO incompatibility due to immunoglobulin G anti-B antibodies presenting with severe fetal anaemia. Transfus Med 15: 57-60.
- Haque KM, Rahman M (2000) An unusual case of ABO-haemolytic disease of the newborn. Bangladesh Med Res Counc Bull 26: 61-64.
- Goraya J, Basu S, Sodhi P, Mehta S (2001) Unusually severe ABO hemolytic disease of newborn. Indian J Pediatr 68: 285-286.
- Marwaha N, Dhawan Hk, Thakral B, Kaur R, Basu S, et al. (2009) Severe ABO hemolytic disease of newborn with a positive direct antiglobulin test. Indian J Pathol Microbiol 52: 292.
- Lambin P, Debbia M, Puillandre P, Brossard Y (2002) IgG1 and IgG3 anti-D in maternal serum and on the RBCs of infants suffering from HDN: relationship with the severity of the disease. Transfusion 42: 1537-1546.
- Quinley ED (1993) Haemolytic disease of the newborn. In: Immunohaematology: Principles and Practice. Lippincott, New York City, USA. Pg no: 277-308.
- Qureshi H, Massey E, Kirwan D, Davies T, Robson S, et al. (2014) BCSH guideline for the use of anti-D immunoglobulin for the prevention of haemolytic disease of the fetus and newborn. Transfus Med 24: 8-20.
- Liu KL, Li N, Li BJ, Peng JY, Shu XW, et al. (2009) [Clinical significance of anti-D IgG screening and titer detection in 286 RhD negative pregnant women]. Zhongguo Dang Dai Er Ke Za Zhi 11: 185-187.
- Lee SM, Im SJ, Park SE, Lee EY, Kim HH (2007) [A case of severe hemolytic disease of the newborn due to anti-Dia antibody]. Korean J Lab Med 27: 373-376.
- Kim WD, Lee YH (2006) A fatal case of severe hemolytic disease of newborn associated with anti-Jkb. J Korean Med Sci 21: 151-154.
- Gao XY, Huang H, Li LD (2009) [Hemolytic disease of neonates due to anti-M: report of one case and review of reports of 21 cases]. Zhonghua Er Ke Za Zhi 47: 648-652.
- Thakral B, Agrawal SK, Dhawan HK, Saluja K, Dutta S, et al. (2007) First report from India of haemolytic disease of newborn by anti-c and anti-E in Rh (D) positive mothers. Hematology 12: 377-380.
- Delaunay J (2002) Molecular basis of red cell membrane disorders. Acta Haematol 108: 210-218.
- Kaplan M (2001) Genetic interactions in the pathogenesis of neonatal hyperbilirubinemia: Gilbert’s Syndrome and glucose-6-phosphate dehydrogenase deficiency. J Perinatol 21 30-39.
- Kaplan M, Hammerman C (2002) Glucose-6-phosphate dehydrogenase deficiency: a potential source of severe neonatal hyperbilirubinaemia and kernicterus. Semin Neonatol 7: 121-128.
- Zanella A, Bianchi P (2000) Red cell pyruvate kinase deficiency: from genetics to clinical manifestations. Baillieres Best Pract Res Clin Haematol 13: 57-81.
- Sohan K, Billington M, Pamphilon D, Goulden N, Kyle P (2002) Normal growth and development following in utero diagnosis and treatment of homozygous alpha-thalassaemia. BJOG 109: 1308-1310.
- Davies SC, Cronin E, Gill M, Greengross P, Hickman M, et al. (2000) Screening for sickle cell disease and thalassaemia: a systematic review with supplementary research. Health Technol Assess 4: 1-99.
- van der Schoot CE, Tax GH, Rijnders RJ, de Haas M, Christiaens GC (2003) Prenatal typing of Rh and Kell blood group system antigens: the edge of a watershed. Transfus Med Rev 17: 31-44.
- Singleton BK, Green CA, Avent ND, Martin PG, Smart E, et al. (2000) The presence of an RHD pseudogene containing a 37 base pair duplication and a nonsense mutation in africans with the Rh D-negative blood group phenotype. Blood 95: 12-18.
- Flegel WA (2011) Molecular genetics and clinical applications for RH. Transfus Apher Sci 44: 81-91.
- Moise KJ (2008) Hemolytic disease of the fetus and newborn. In: Greene MF, Creasy RK, Resnik R, Iams JD, Lockwood CJ et al. (eds.). Maternal-fetal Medicine: Principles and Practice, (6th edn), Saunders, Philadelphia, USA. Pg no: 477-503.
- Kaplan M, Na'amad M, Kenan A, Rudensky B, Hammerman C, et al. (2009) Failure to predict hemolysis and hyperbilirubinemia by IgG subclass in blood group A or B infants born to group O mothers. Pediatrics 123: 132-137.
- Luchtman-Jones L, Schwartz AL, Wilson DB (2006) The blood and hematopoietic system. In: Fanaroff AA, Martin RJ (eds.). Neonatal-Perinatal Medicine-Diseases of the Fetus and Infant, (8th edn), Mosby, Maryland Heights, USA. Pg no: 1287-1356.
- Newman TB, Liljestrand P, Jeremy RJ, Ferriero DM, Wu YW, et al. (2006) Outcomes among newborns with total serum bilirubin levels of 25 mg per deciliter or more. N Engl J Med 354: 1889-1900.
- Wu KH, Chu SL, Chang JG, Shih MC, Peng CT (2003) Haemolytic disease of the newborn due to maternal irregular antibodies in the Chinese population in Taiwan. Transfus Med 13: 311-314.
- Koelewijn JM, Vrijkotte TG, van der Schoot CE, Bonsel GJ, de Haas M (2008) Effect of screening for red cell antibodies, other than anti-D, to detect hemolytic disease of the fetus and newborn: a population study in the Netherlands. Transfusion 48: 941-952.
- Koelewijn JM, Vrijkotte TG, de Haas M, van der Schoot CE, Bonsel GJ (2009) Risk factors for the presence of non-rhesus D red blood cell antibodies in pregnancy. BJOG 116: 655-664.
- The Royal Australian and New Zealand College of Obstetricians and Gynaecologists (2007) Guidelines for blood grouping and antibody screening in the antenatal and perinatal setting. Australian and New Zealand Society of Blood Transfusion Ltd, (3rd edn), Sydney, Australia.
- Finning K, Martin P, Summers J, Daniels G (2007) Fetal genotyping for the K (Kell) and Rh C, c, and E blood groups on cell-free fetal DNA in maternal plasma. Transfusion 47: 2126-2133.
- Daniels G, Finning K, Martin P, Soothill P (2004) Fetal blood group genotyping from DNA from maternal plasma: an important advance in the management and prevention of hemolytic disease of the fetus and newborn. Vox Sang 87: 225-232.
- van Rossum HH, de Kraa N, Thomas M, Holleboom CAG, Castel A, van Rossum AP (2015) Comparison of the direct antiglobulin test and the eluate technique for diagnosing haemolytic disease of the newborn. Practical Laboratory Medicine 3: 17-22.
- Dinesh D (2005) Review of positive direct antiglobulin tests found on cord blood sampling. J Paediatr Child Health 41: 504-507.
- Koenig JM (2000) Evaluation and treatment of erythroblastosis fetalis in the neonate. In: Christensen RD (ed.). Hematologic Problems of the Neonate. Saunders, Philadelphia, USA. Pg no: 185-207.
- Christensen RD, Henry E (2010) Hereditary spherocytosis in neonates with hyperbilirubinemia. Pediatrics 125: 120-125.
- Murray NA, Roberts IA (2007) Haemolytic disease of the newborn. Arch Dis Child Fetal Neonatal Ed 92: 83-88.
- Bakkeheim E, Bergerud U, Schmidt-Melbye AC, Akkök CA, Liestøl K, et al. (2009) Maternal IgG anti-A and anti-B titres predict outcome in ABO-incompatibility in the neonate. Acta Paediatr 98: 1896-1901.
- Liumbruno GM, D'Alessandro A, Rea F, Piccinini V, Catalano L, et al. (2010) The role of antenatal immunoprophylaxis in the prevention of maternal-foetal anti-Rh(D) alloimmunisation. Blood Transfus 8: 8-16.
- Mari G, Deter RL, Carpenter RL, et al (2000) Noninvasive diagnosis by Doppler ultrasonography of fetal anemia due to maternal red-cell alloimmunization. Collaborative Group for Doppler Assessment of the Blood Velocity in Anemic Fetuses. N Engl J Med 342: 9-14.
- van Kamp IL, Klumper FJ, Meerman RH, Oepkes D, Scherjon SA, et al. (2004) Treatment of fetal anemia due to red-cell alloimmunization with intrauterine transfusions in the Netherlands, 1988-1999. Acta Obstet Gynecol Scand 83: 731-737.
- Booker W, Fox NS, Gupta S, Carroll R, Saltzman DH, et al. (2015) Antenatal Surveillance in Twin Pregnancies Using the Biophysical Profile. J Ultrasound Med 34: 2071-2075.
- Payne BA, Kyle PM, Lim K, Lisonkova S, Magee LA, et al. (2013) An assessment of predictive value of the biophysical profile in women with preeclampsia using data from the fullPIERS database. Pregnancy Hypertens 3: 166-171.
- Oyelese Y, Vintzileos AM (2011) The uses and limitations of the fetal biophysical profile. Clin Perinatol 38: 47-64.
- Kehl S, Schelkle A, Thomas A, Puhl A, Meqdad K, et al. (2015) Single deepest vertical pocket or amniotic fluid index as evaluation test for preventing adverse pregnancy outcome (SAFE trial): a multicentre, open-label randomised controlled trial. Ultrasound Obstet Gynecol 47: 674-679.
- Gearhart PA (2015) Ultrasonography in Biophysical Profile. Medscape, New York, USA.
- Petrozella LN, Dashe JS, McIntire DD, Leveno KJ (2011) Clinical significance of borderline amniotic fluid index and oligohydramnios in preterm pregnancy. Obstet Gynecol 117: 338-342.
- Sande JA, Ioannou C, Sarris I, Ohuma EO, Papageorghiou AT (2015) Reproducibility of measuring amniotic fluid index and single deepest vertical pool throughout gestation. Prenat Diagn 35: 434-439.
- Yaseen H, Khalaf M, Rashid N. et al (2005) Does prophylactic phototherapy prevent hyperbilirubinemia inneonates with ABO incompatibility and positive Coombs’ test? J Perinatol 25: 590-594.
- McDonagh AF (2001) Phototherapy: from ancient Egypt to the new millennium. J Perinatol 21: 7-12.
- Wagle S (2016) Hemolytic Disease of Newborn Medication. Medscape, New York, USA.
- Delaney M, Matthews DC (2015) Hemolytic disease of the fetus and newborn: managing the mother, fetus, and newborn. Hematology Am Soc Hematol Educ Program 2015: 146-151.
- Harkness UF, Spinnato JA (2004) Prevention and management of RhD isoimmunization. Clin Perinatol 31: 721-742.
- Thaithumyanon P, Visutiratmanee C (2002) Double phototherapy in jaundiced term infants with hemolysis. J Med Assoc Thai 85: 1176-1181.
- Vreman HJ, Wong RJ, Stevenson DK (2004) Phototherapy: current methods and future directions. Semin Perinatol 28: 326-333.
- Seidman DS, Moise J, Ergaz Z et al. (2003) A prospective randomized controlled study of phototherapy using blue and blue-green light-emitting devices, and conventional halogen-quartz phototherapy. J Perinatol 23: 123-127.
- Gottstein R, Cooke RW (2003) Systematic review of intravenous immunoglobulin in haemolytic disease of the newborn. Arch Dis Child Fetal Neonatal Ed 88: 6-10.
- Elalfy MS, Elbarbary NS, Abaza HW (2011) Early intravenous immunoglobin (two-dose regimen) in the management of severe Rh hemolytic disease of newborn--a prospective randomized controlled trial. Eur J Pediatr 170: 461-467.
- Smits-Wintjens VE, Walther FJ, Rath ME, Lindenburg IT, te Pas AB, et al. (2011) Intravenous immunoglobulin in neonates with rhesus hemolytic disease: a randomized controlled trial. Pediatrics 127: 680-686.
- Kappas A (2004) A method for interdicting the development of severe jaundice in newborns by inhibiting the production of bilirubin. Pediatrics 113: 119-123.
- Miqdad AM, Abdelbasit OB, Shaheed MM, Seidahmed MZ, Abomelah AM, et al. (2004) Intravenous immunoglobulin (IVIG) therapy for significant hyperbilirubinemia in ABO hemolytic disease of the newborn. J Matern Fetal Neonatal Med 16: 163-166.
- Mukhopadhyay K, Murki S, Narang A, Dutta S (2003) Intravenous immunoglobulins in rhesus hemolytic disease. Indian J Pediatr 70: 697-699.
Citation: Esan AJ (2016) Hemolytic Disorders of the Newborn, Current Methods of Diagnosis and Treatment: A Review Study. J Hematol Blood Transfus Disord 3: 008.
Copyright: © 2016 Esan AJ, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.