Immune Response in Various Diffuse Parenchymal Lung Disorders (DPLDs)
*Corresponding Author(s):
Syed Zulkharnain TousheedDepartment Of Pulmonology, Narayana Health, Bangalore, India
Tel:+91 8071222142,
Email:syed.tousheed@gmail.com
Abstract
Lung has one of the largest surface areas. It has to bear the brunt of infinite insults both from the airway and vascular end. The inhaled particles depending upon the size are cleared either by the luminal or the alveolar or the interstitial macrophages. The alveolar and interstitial macrophages clear the engulfed particles via the lymphatic system, which depends upon the blood perfusion. Most of the particles from the lower and middle lobes are cleared successfully, but the particle engulfed by macrophages in the upper lobe get trapped in the interstitium because of poor lymphatic supply. This leads to systemic immune response and further formation of a granuloma and fibrosis. Depending upon on the type of immune pathway, the granulomas are either compact as in Th2 pathway or lose granulomas as in Th1 pathway. In some situations due to extreme peripheral deposition of particulate matters as in smoking or due to alveolar damage, the mesenchymal cells which reside in the broncho-alveolar duct junctions can get stimulated. The mesenchymal cells includes the resident fibroblast, lipofibroblast and endothelial pericytes. Stimulation of these cells results in formation of myofibroblast, which results in dysregulated fibrosis as in idiopathic pulmonary fibrosis.
Lung, on the vascular end while filtering substances in the circulation can get exposed to their harmful effects, which can cause local damage. Due to the influence of gravity, the lower lobes of the lungs are more perfused. The toxins from vascular side get in contact with the lower lobe relatively more and hence causes more damage in the lower lobes. Also in the periphery of the lower lobe the toxins have a longer distance to travel at a relatively slower pace. The greater transit time for immune mediated cells, complexes or malignant cells makes the peripheral part of lung more vulnerable for immune mediated damage.
Hence the involvement of different part of the lung depends on the type of injury, etiology and the duration of the injury. Inhalational injury most of the time involves the upper lobe except in smoking, while the insult from vascular side leads to the injury of lower lobes. Nevertheless, any injury either from airway or from the vascular side causes an inflammatory damage. The injury either can repair (completely or partially) or it can regenerate the damaged alveoli or it can form a fibrous tissue. These different phases of response by the lung parenchyma to a particular injury can give rise to different patterns on radiology and histopathology. Though the insults to the lung parenchyma may be infinite, the response patterns are finite. Different group of disorders with similar response to different types of etiologies are grouped under Diffuse Parenchymal Lung Disease (DPLD). It is important to know the predominant pattern in a particular disease because modality of treatment we choose depends on this.
Keywords
Hypersensitivity pneumonitis; Idiopathic pulmonary fibrosis; Lung disorders; Parenchymal lung disease; Pneumonia
DISCUSSION
Lung as a solid organ has one of the largest surface areas .It is said that the surface area of lung is equal to the area of a lawn tennis court [1,2]. This enables the respiratory system to have a tremendous amount of reserve for gas exchange across the alveolo-capillary membrane. Because of this enormous surface area, it gets exposed to infinite number of insults both from the external and internal environment. Like skin it acts as the primary defense for the external particulate matters which are inhaled [3]. On the other side it act as a check post between venous and systemic circulation. It filters substances like clots, fibrin clumps, and other endogenous chemicals and exogenous materials from entering the systemic circulation [1,2].
Larger particles inhaled are engulfed by the luminal airway macrophages and are expelled out via the mucociliary escalator [4]. If the particle size is small enough to reach the alveoli they are engulfed by the Pulmonary Alveolar Macrophages (PAMs) and are cleared by the lymphatic system [3-5]. In some situations the particles cannot be cleared by pulmonary alveolar macrophages, hence these antigen may be internalized by the type 1 pneumocytes and hence enter the interstitium. The Interstitial macrophages engulf these antigens from there and try to clear them via the lymphatics [4].
But the lymphatic clearance is a function of the lymphatic flow, perfusion and respiratory excursion, all of which are relatively deceased in the upper lung zones. Hence the engulfed antigen remains for a longer time in the interstitium in upper lobes [6]. The interstitial macrophages containing the antigen in the upper lobes recruits circulating macrophages, lymphocytes, fibroblast by producing cytokines. The interaction between macrophages and T- lymphocytes are mediated by cytokines which leads to formation of a granuloma. Depending on the type of cytokine released, the granuloma can be either loose (CD8 mediated) or compact (CD4 mediated) [7,8]. Granuloma is the penultimate line of pulmonary defense. The CD4 T lymphocytes in a compact granuloma attracts more fibroblast which would lead to parenchymal fibrosis [8].
When particles like silica and asbestos are inhaled, they are extremely toxic to cells. These particles cannot be broken down by lysozymal enzymes present in the PAMs. Thus, the engulfed particles can act as a mild persistent irritants leading to chronic inflammation. When the dose of the inhaled particle is very high they can destroy the host macrophages and cause spillover of the lysozymes to the exterior. This would lead to significant inflammation and fibrosis [5].
Most of the inhaled agents cause upper lobe predominant DPLDs except smoking. Tobacco smoke, being a complex mixture of gaseous and particulate phases with several toxic chemicals seems to behave differently from other particles. Most of the known DPLDs caused by smoking are lower lobe predominant [9]. This pattern of involvement may be due to size behavior of the particulate matters in smoke stimulating the different types of macrophages and mesenchymal cells [4,10,11].
Lung, on the vascular end while filtering many substances in the circulation can get exposed to their harmful effects, which can cause local damage in the form of inflammation and fibrosis (Figure 1) [1].
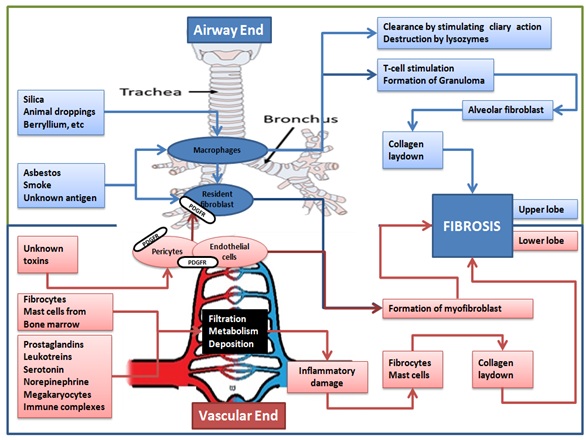 Figure 1: Showing lung defence and response to various insults.
Figure 1: Showing lung defence and response to various insults.
Since lung act as a check post, most of the venous blood has to pass through the pulmonary circulation. Substances like amines, peptides, arachidonic acid derivatives pass through the pulmonary circulation. Few of them have a silent transit, neither they get activated nor inactivated, but a few of them get inactivated during their transit [1]. Either excessive load of these substances or inability of the lung to inactivate these substances can cause immune damage to the lungs. Due to the influence of gravity the lower lobes of the lungs are more perfused. The toxins from vascular side get in contact with the lower lobe relatively more and hence causes more damage in the lower lobes. Also, in the periphery of the lower lobe the toxins have a longer distance to travel at a relatively slower pace. This leads to greater transit time for immune mediated cells, complexes, or malignant cells in the pulmonary circulation. The enormous surface area along with the increased transit time increases the interaction between toxins in the blood and the peripheral lung parenchyma. This makes it more likely for toxic substances to deposit and cause inflammatory damage in the lower and peripheral part of lung. Hence interstitial pneumonias secondary to autoimmune disorders and drug-induced toxicity, which are associated with circulating agents & immune complexes are lower lobe and peripheral dominant [6].
The large surface area and the narrow diameter of pulmonary capillaries make the immune complexes and inflammatory cells like fibrocytes and monocytes to get trapped [12,13]. This triggers local damage; damaged tissues expose previously sequestered antigens to the immune system causing progression of the damage locally. This may explain the reason for the frequent involvement of the lung in systemic immune disorders even when the other clinical features of the primary connective tissue disorder may be minimal or absent, as in Idiopathic Pneumonia with Autoimmune features (IPAF). Pulmonary cells themselves secrete few substances during stressful situation like prostaglandins, leukotreins, serotonin, megakaryocytes etc, which can themselves cause inflammation and fibrosis by recruiting more cells [1] (Figure 1).
Hence the involvement of different part of the lung depends on the type of injury (inhalation or vascular), etiology (autoimmune, idiopathic, or infectious) and the duration of the injury (acute, chronic).
Inhalational injury most of the time involves the upper lobe except smoking, while the insult from vascular side leads to the injury of lower lobes. Nevertheless, any Injury either from airway or from the vascular side initiates an inflammatory response, which can either heal completely or partially (on its own or with treatment) or it can progress to form a fibrous tissue [14].
Fibrosis is either an attempt to wall off the antigen in some conditions or an aberrant healing in a few. Ultimately fibrosis leads to the distortion and destruction of surrounding normal alveolar architecture resulting in the clinical consequences typical of DPLDs like cough ( due to stimulation of cough receptors in the alveoli) and breathlessness (due to ventilation perfusion mismatch, impaired oxygen diffusion as well as increased work of breathing due to the distorted, thickened and stiff interstitium).
These different phases of response by the lung parenchyma to a particular injury can give rise to different patterns on radiology and histopathology. Though the insults to the lung parenchyma may be infinite, the response patterns are finite. Different group of disorders with similar response to different types of etiologies are grouped under Diffuse Parenchymal Lung Disease (DPLD). DPLDs are classified based on etiology and different patterns [15-17] (Figure 2).
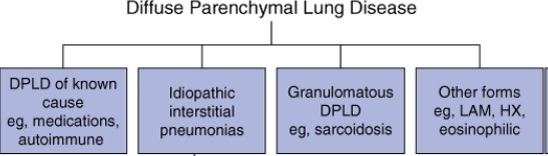 Figure 2: Showing the classification of DPLDS.
Figure 2: Showing the classification of DPLDS.
Each etiological factor can give rise to more than one pattern (Figure 3) [18] and these patterns are defined based on presence of consolidation, inflammation and fibrosis, predominantly on a CT scan. It is important to know the predominant pattern in a particular disease because modality of treatment we choose depends on this [18].
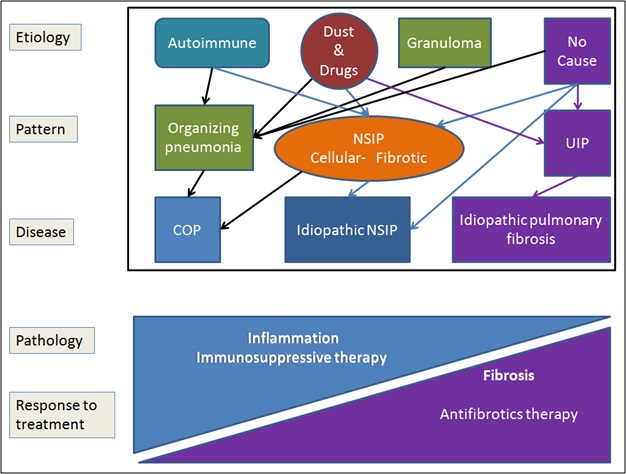 Figure 3: Showing different etiologies giving rise to different patterns of DPLDs.
Figure 3: Showing different etiologies giving rise to different patterns of DPLDs.
MAJOR PATTERNS OF DPLD
Granulomatous patterns
Common DPLDs which present with granulomas are Sarcoidosis and Hypersensitivity Pneumonitis (HP) etc.
The Granulomatous diseases in chronic HP and Sarcoidosis have predilection for upper lobes. The inhaled antigens are engulfed by resident alveolar macrophages which fail to clear the antigen on its own. The macrophages entrap these antigens and triggera T cell response. The cytokines and chemokines released by the activated immune cells attract more inflammatory cells to the site. Different immune-regulatory cells accumulate and organize themselves to form a granuloma, either to eliminate or restrict the antigen.
The morphology of granuloma depends upon the cytokine released by the inflammatory cells, which in turn depends on the intention of the immune system weather to eliminate or restrict the antigen. To restrict the antigen, immune system adapts to the Th2 pathway. The Tumour Necrosis Alfa (TNF-Alfa) being the predominant cytokine shifts the balance towards a Th2 pathway which leads to formation of compact granuloma, rich in CD4 lymphocytes as in Sarcoidosis [7]. In acute and subacute Hypersensitivity pneumonitis the granulomas are dominated by CD8 lymphocytes. CD8 lymphoctes are the cytotoxic cells which try to eliminate the antigen presented to them. Most of the time they are successful in clearing the inciting antigen via the Th1 cytokine pathway. Interferon Gamma is the most important Th1 cytokine, which inhibits fibrosis [19]. Hence the granulomas in acute and subacute HP are loose granulomas with a low CD4/CD8 ratio and lacking fibrotic cells [8]. If the antigen exposure persists for a long time, the CD8 cells get exhausted and fail to eliminate the antigen. The pathway then shifts from Th1 to Th2 (CD8 to CD4) pathway. The CD4/CD8 ratio starts increasing as chronic HP sets in. Interleukin-4 & interleukin -13 are more pro-fibrotic cytokines, which are a part of Th2 pathway [20,21]. When the immune system fails to eliminate an antigen via Th1 pathway (CD8 predominant), it adapts a more conservative Th2 (CD4 predominant) strategy by fencing (fibrosis) the area around antigen.
Sarcoidosis
Sarcoidosis predominantly involves intrathoracic lymphadenopathy with or without egg shell calcification (hilar, mediastinal, paraaortic, subcarinal) with predilection for upper and mid lung zones. Perihilar lymphatics nodules (fissural), confluent alveolar opacities, ground glass opacities, interlobular septal thickening & subpleural linear opacities may be seen [22,23].
HYPERSENSITIVITY PNEUMONITIS (HP)
Acute and subacute HP
The inflammation in this pattern almost exclusively involves distal airways, alveoli and interstitium around these airways. In acute HP, predominant inflammatory cells are neutrophils, while in sub-acute HP it is characterized by the lymphocytic infiltration with ill formed granulomas [24]. Granulomas are seen in the bronchiolar wall and alveolar ducts. These granuloma are smaller than those seen in sarcoidosis and they lack fibrosis around as in Sarcoidosis. HRCT shows diffuse ground glass opacities, peribronchovascular nodules in the middle and lower lobes. Minimal fibrosis is noted [25].
Chronic HP
The characteristic feature is bronchiolo-centric inflammation with poorly formed non-necrotizing granulomas. Bronchiolitis can be either cellular, follicular, or obliterans type. Other important finding is the Peribronchiolar metaplasia with Airway-Centered Interstitial Fibrosis ACIF) [26,27].
Centrilobular fibrosis in HP is the bridging fibrosis (fibrosis between the respiratory bronchioles and the interlobular septa or with adjacent respiratory bronchioles or with subpleural areas). Sometimes an organizing pneumonia can also be seen in chronic HP. Presence of centrilobular nodules in imaging is one of the differentiating feature from other patterns like UIP and NSIP [28].
Cryptogenic organizing pneumonia
When the usual process of resolution in a pneumonia has failed and there is an organization of the inflammatory exudates in the alveoli resulting in fibrosis is called as Organizing Pneumonia (OP) [29,30]. This can be due to many causes. The inciting agent can be either from vascular side or the airway side. When the cause remains unknown despite extensive evaluation it is termed as Cryptogenic Organizing Pneumonia (COP) [29,30].
The typical pattern is a patchy organizing pneumonia involving alveolarducts and alveoli and may be with bronchiolar intraluminal polyps (mason bodies).Some of them showing marked interstitial inflammation. On imaging patchy migratory consolidation are seen in the subpleural or peribronchial region. Ground-glass opacity, perilobular opacities and reversed halo (or atoll) sign may also be seen which would help in the diagnosis of organizing pneumonia [17,31-35].
Non-Specific Interstitial Pneumonitis (NSIP) patterns
Inflammation and fibrosis which are uniform (temporal homogeneity) in nature are a feature of idiopathic NSIP. The inflammatory cells are predominantly lymphocytes and plasma cells.
Inflammation or fibrosis in NSIP predominantly involves lower lung and it can either be diffuse or peripheral. Reticular abnormalities, traction bronchiectasis, lobar volume loss can also be seen. Ground-glass attenuation suggesting active inflammation is a distinguishing feature from UIP. Sub-pleural sparing and peribronchial thickening can also be seen. NSIP patterns can be seen in Hypersensitivity Pneumonitis or connective tissue disorders like Scleroderma. If the cause is unknown it is grouped as idiopathic NSIP (iNSIP) [17,36]. The injury for this pattern again can be from either vascular side when associated with connective tissue disorders or from inhalation side when associated with HP. In an idiopathic NSIP the inciting agent logically should be from vascular end as it involves lower lobes .Since the predominant cells are lymphocytes and plasma cells systemic immune response is more pronounced in this pattern. Infact, few authors consider iNSIP as lung involvement of a undifferentiated connective tissue disorder. Various terminologies like lung dominant connective tissue disease was used to describe this entity. Later this disorder was better recognized and defined as Interstitial Pneumonia with Autoimmune Features (IPAF) [37]. Studies have found that iNSIP patients would have some autoimmune clinical features like arthralgia, fever, myositis etc [38]. Presence of lymphoplasmacytic infiltrates which usually are a feature of autoimmune process strengthens the hypothesis [38]. As described earlier the transit time is more in the lower lobe peripheries, the chances of peripheral involvement of lung is more common when NSIP pattern is associated with connective tissue disorders and malignancies [6]. While peribronchial thickening and diffuse involvement could be seen in NSIP secondary to HP or any inhalational injury. Also the type of fibrosis in iNSIP is different from UIP (Usual Interstitial Pneumonia). Fibrosis in non UIP patterns is probably by the alveolar and bone marrow derived fibroblast [39] unlike UIP where resident fibroblast, lipofibroblast and pericytes play a significant role [39].
Usual interstitial pneumonia (UIP) pattern
This pattern contains more of fibrosis than inflammation. The fibrotic areas are predominantly located in the lower lobes, peripheral zones of secondary lobules and along with perivenular areas located between primary lobules. Usually the injured foci are seen around these fibrotic areas. The characteristic feature of this pattern is the Fibroblastic Foci (FF), which is composed of fibroblasts and myofibroblasts [40]. These fibroblasts and myofibroblast are derived from the subepithelial resident fibroblast, lipofibroblast and interendothelial pericytes. These cells are important to maintain the airway epithelial and vascular integrity [11]. These cells play a significant role in the development of the lung during embryonic stage by their interaction with the epithelial and the endothelial cells respectively. Whenever there is an injury either from vascular end or from airway end, the repair mechanisms in the lungs kick in, bringing back these cells in action. The entire process of repair mechanism in an adult lung is considered as restarting of the development process. The multipotent mesenchymal cells which have high regenerative capacity are usually resting in the terminal most part of the airways called as Bronchoalveolar Duct Junctions (BADJ) [41]. There is a complex interaction between the mesenchymal cells (resident fibroblast & subset of pericytes), epithelial and endothelial cells, similar to the development process when there is an injury. The interaction between these cells leads to either regeneration or remodeling of the injured lung parenchyma. If the environment tips towards remodeling, fibrosis sets in rather than re-epithelialization. Idiopathic Pulmonary Fibrosis (IPF) with UIP pattern is one such condition where there is remodeling with an aberrant wound healing ending up with fibrosis [42-44]. The peripheral location for the multipotent mesenchymal cells could be the reason for the subpleural fibrosis in UIP pattern. These mesenchymal cells express Platelet Derived Growth Factor Receptors (PDGFR) on them in abundance. Through the PDGF ligands, these cells maintain homeostasis and integrity of airway cells and vascular endothelium. During the tissue injury there is production of PDGF to facilitate healing by the resident fibroblast, lipofibroblast and pericytes [39]. In conditions like IPF, there is unchecked proliferation of these cells and transition of these cells to myofibroblasts [39]. These myofibroblasts causes excess production of extra cellular matrix resulting in fibrosis [45]. PDGF responsible for the remodeling and fibrosis could be the reason for digital clubbing commonly seen in IPF [13].
Hence targeting the PDGFR present on these mesenchymal cells responsible for fibrosis using molecules like Nintedanib would be more logical in IPF than using regular immunosuppressants which act on adaptive immunity [11].
SMOKING RELATED DPLDS
Inhalation of tobacco and its products stimulates both innate immunity and adaptive immunity. Innate immunity predominantly includes macrophages, dendritic cells and mesenchymal cells. While adaptive immunity consists of lymphocyte response, which could either be B cell or T cell response [46]. Depending on the size of the particulate matter in the smoke, either airway luminal or the alveolar macrophages are stimulated. Larger sized particulate matters are engulfed by airway luminal macrophages and are expelled via the mucociliary apparatus. While the respirable sized particulate matter in smoke are engulfed by alveolar macrophages, which usually mounts a mild immune response [4,11]. The macrophages secrete various chemokines and cytokines which enhances the adaptive immunity. The macrophages can also stimulate the formation of Bronchus-Associated Lymphoid Tissue (BALT) and hence initiating a lymphocytic inflammation. When the burden of the particulate matter is significant the interstitial macrophages may get involved and help in mounting intense systemic inflammation. This would recruit more inflammatory cells like neutrophils, monocytes and fibrocytes [4]. The ultrafine smoke particles get deposited in the peripheral most part of the lung in high concentration [10] which could stimulate the subepithelial fibroblast directly or indirectly. These resident fibroblast lay down the extra cellular matrix in patches in the periphery [11]. Different patterns of DPLDs are associated with smoking related injury. The most important ones are IPF (UIP), Respiratory Bronchiolitis associated Interstitial Lung Disease (RB-ILD), Desquamative Interstitial Pneumonia and Pulmonary Langerhan cell Histiocytosis (PLCH) (Tables 1 -3, figure 4) [9].
|
|
Sarcoid |
Acute /sub acute HP |
Chronic HP |
|
Pathology |
Upper lobe predominant Fibrosis Both periphery & peribronchovascular areas involved Non caseating compact granuloma CD4 lymphocytes predominant |
Middle &lower lobe predominant inflammation Non caseating lose granulomas CD8 lymphocyte predominant |
Upper lobe predominant fibrosis Bridging fibrosis and airway centered interstitial fibrosis (ACIF) noted CD4 lymphocyte predominant |
|
HRCT |
Upper lobe fibrosis Fissural nodules Hilar and mediastinal lymphadenopathy with or without eggshell calcification |
Diffuse GGO Peribronchovascular nodules Lower lobe predominant disease |
Upper lobe predominant fibrosis Traction bronchiectasis Peribronchovascular involvement |
|
Response to steroids |
Good |
Good |
Fair |
|
Recovery |
Good |
Good |
Fair |
Table 1: Showing features of granulomatous DPLDs [6-8,22-28].
|
|
UIP (IPF) |
iNSIP |
COP |
|
Pathology |
Fibrosis in lower lobes & periphery Fibroblastic Foci (FF) Heterogenous, patchy, inflammation seen Minimal inflammatory cells |
Fibrosis in lower lobes No FF but fibroblast seen Homogenous inflammation Lymphocytes & plasma cells seen |
Intra luminal fibrotic polyps in the alveoli Peribronchial, Patchy & peripheral consolidation Neutrophils, lymphocytes & eosinophils seen |
|
CT thorax |
Honey combing in lower lobes & periphery Fibrosis & Traction bronchiectasis No GGO |
Lower lobe fibrosis with Sub pleural sparing Traction bronchiectasis GGO can be seen |
Migratory patchy consolidations both peribronchial & peripheral Reverse Halo(Atoll) sign GGO seen |
|
Response to steroids |
No |
Fair |
Good |
|
Recovery |
NO |
Partial |
Good |
Table 2: Showing features of IIPs [17,30,31,36].
|
|
PLCH |
RB-ILD |
DIP |
|
Pathology |
· Langerhans cells containing nodules · Intervening normal lung · Cystic lesions · Fibrosis in peribronchiolar area |
· Pigmented macrophages · Respiratory bronchiolitis · Peribronchiolar interstitial inflammation · Minimal fibrosis |
· Pigmented macrophages · Mild interstitial inflammation · Diffuse fibrosis · Lung architecture maintained |
|
CT Thorax |
Nodules Cysts |
Patchy GGO Nodules |
Lower lobe predominant GGO |
|
Response to steroids |
Fair |
Good |
Good |
|
Recovery |
Yes |
yes |
Yes |
Table 3: Smoking related DPLDs other than IPF [9].
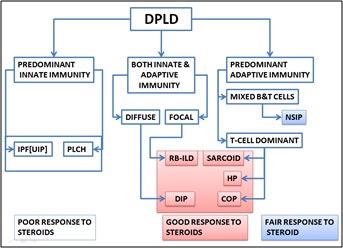 Figure 4: Showing classification major DPLDs based on type of Immunity involved.
Figure 4: Showing classification major DPLDs based on type of Immunity involved.
SUMMARY
- • DPDLs are a group of disorders with different patterns on imaging and histopathology
- • Type of pattern in a DPLD depends on the route of the insult (inhalation/circulation), etiology (autoimmune/infection/smoke), duration of insult (acute/chronic), physical properties of the agent (small/large) or the type of immune response the agent stimulates(innate/adaptive/combined)
- • During a response to the alveolar injury, the process of development restarts again
- • In conditions like IPF, process of remodeling predominantes rather than regeneration during healing. Hence fibrosis replaces normal alveolarization.
- • NSIP behaves like a lung dominant CTD-ILD where lung might act as an auto antigen
- • Most of the smoking related DPLs are lower lobe predominant unlike other inhalation agents which causes upper lobe predominant diseases.
- • Granuloma formation is a penultimate measure by the immune sytem to contain the antigen which are not cleared by the macrophages.
- • COP pattern is seen when the usual process of resolution in a pneumonia has failed and there is an organisation of the inflammatory exudate in the alveoli resulting in fibrosis.
- • It is important to know the pattern of DPLD and the type of immune response by the body to chose an optimum treatment.
- • Diseases like IPF which are predominantly innate immune response may not respond to steroids. Anti-growth factors like VEGF, PDGF and TGF-β may be more suitable in this condition.
REFERENCES
- Joseph D, Puttaswamy RK, Krovvidi H (2013) Non-respiratory functions of the lung. Critical Care and Pain 13: 98-102.
- Levitzky MG (2003) Pulmonary Physiology (6th edn). McGraw-Hill, New York, USA.
- Nicod LP (1999) Pulmonary defence mechanisms. Respiration International Review of Thoracic Diseases 66: 2-11.
- Lehnert BE (1992) Pulmonary and thoracic macrophage subpopulations and clearance of particles from the lung. Environmental Health Perspectives 97: 17-46.
- Parks CG, Conrad K, Cooper GS (1999) Occupational exposure to crystalline silica and autoimmune disease. Environmental Health Perspectives 5: 793-802.
- Nemec SF, Bankier AA, Eisenberg RL (2013) Upper lobe-predominant diseases of the lung. AJR American journal of roentgenology 200: 222-237.
- Broos CE, van Nimwegen M, Hoogsteden HC, Hendriks RW, Kool M, et al. (2013) Granuloma formation in pulmonary sarcoidosis. Frontiers in Immunology 4: 437.
- Barrera L, Mendoza F, Zuñiga J, Estrada A, Zamora AC, et al. (2008) Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. American Journal of Respiratory and Critical Care Medicine 177: 44-55.
- Ryu JH, Colby TV, Hartman TE, Vassallo R (2001) Smoking-related interstitial lung diseases: A concise review. The European Respiratory Journal 17: 122-132.
- Muller WJ, Hess GD, Scherer PW (1990) A model of cigarette smoke particle deposition. American Industrial Hygiene Association Journal 51: 245-256.
- Barron L, Gharib SA, Duffield JS (2016) Lung pericytes and resident fibroblasts: Busy multitaskers. The American Journal of Pathology 186: 2519-2531.
- Bagnato G, Harari S (2015) Cellular interactions in the pathogenesis of interstitial lung diseases. European Respiratory Review 24: 102-114.
- Dickinson CJ, Martin JF (1987) Megakaryocytes and platelet clumps as the cause of finger clubbing. Lancet 2: 1434-1435.
- Wilson MS, Wynn TA (2009) Pulmonary fibrosis: Pathogenesis, etiology and regulation. Mucosal Immunology 2: 103-121.
- American Thoracic Society, European Respiratory Society (2002) American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 165: 277-304.
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, et al. (2011) An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 183: 788-824.
- Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, et al. (2013) An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 188: 733-748.
- Mikolasch TA, Garthwaite HS, Porter JC (2017) Update in diagnosis and management of interstitial lung disease. Clin Med (Lond) 17: 146-153.
- Sempowski GD, Derdak S, Phipps RP (1996) Interleukin-4 and interferon-gamma discordantly regulate collagen biosynthesis by functionally distinct lung fibroblast subsets. Journal of Cellular Physiology 167: 290-296.
- Liu X, Kohyama T, Wang H, Zhu YK, Wen F-Q, et al. (2002) Th2 cytokine regulation of type I collagen gel contraction mediated by human lung mesenchymal cells. American Journal of Physiology Lung Cellular and Molecular Physiology 282: 1049-1056.
- Wynn TA (2004) Fibrotic disease and the T(H)1/T(H)2 paradigm. Nature Reviews Immunology 4: 583-594.
- National Library of Medicine (1999) Statement on sarcoidosis. Joint Statement of the American Thoracic Society (ATS), the European Respiratory Society (ERS) and the World Association of Sarcoidosis and Other Granulomatous Disorders (WASOG) adopted by the ATS Board of Directors and by the ERS Executive Committee, February 1999. Am J Respir Crit Care Med 160: 736-755.
- Lynch JP (2003) Computed tomographic scanning in sarcoidosis. Semin Respir Crit Care Med 24: 393-418.
- Riario Sforza GG, Marinou A (2017) Hypersensitivity pneumonitis: A complex lung disease. Clinical and molecular allergy 15: 6.
- Anna-Luise K (2006) Katzenstein and Askin’s Surgical Pathology of Non-Neoplastic Lung Disease (4th edn) Elsevier Saunders, Philadelphia, USA. Pg no: 512.
- Pereira CA, Gimenez A, Kuranishi L, Storrer K (2016) Chronic hypersensitivity pneumonitis. Journal of Asthma and Allergy 9: 171-181.
- Kuranishi LT, Leslie KO, Ferreira RG, Coletta EAN, Storrer KM, et al. (2015) Airway-centered interstitial fibrosis: Etiology, clinical findings and prognosis. Respiratory Research 16: 55.
- Silva CIS, Müller NL, Lynch DA, Curran-Everett D, Brown KK, et al. (2008) Chronic hypersensitivity pneumonitis: Differentiation from idiopathic pulmonary fibrosis and nonspecific interstitial pneumonia by using thin-section CT. Radiology 246: 288-297.
- Milne L (1911) Chronic Pneumonia (Including a Discussion of Two Cases of Syphilis of The Lung). The American Journal of the Medical Sciences 142: 408-438.
- Cordier JF (2006) Cryptogenic organising pneumonia. The European Respiratory Journal 28: 422-446.
- Lee JS, Lynch DA, Sharma S, Brown KK, Müller NL (2003) Organizing pneumonia: Prognostic implication of high-resolution computed tomography features. Journal of Computer Assisted Tomography 27: 260-265.
- Lee KS, Kullnig P, Hartman TE, Müller NL (1994) Cryptogenic organizing pneumonia: CT findings in 43 patients. AJR American journal of roentgenology 162: 543-546.
- Müller NL, Staples CA, Miller RR (1990) Bronchiolitis obliterans organizing pneumonia: CT features in 14 patients. AJR American journal of roentgenology 154: 983-987.
- Ujita M, Renzoni EA, Veeraraghavan S, Wells AU, Hansell DM (2004) Organizing pneumonia: perilobular pattern at thin-section CT. Radiology 232: 757-761.
- Kim SJ, Lee KS, Ryu YH, Yoon YC, Choe KO, et al. (2003) Reversed halo sign on high-resolution CT of cryptogenic organizing pneumonia: Diagnostic implications. AJR American journal of roentgenology 180: 1251-1254.
- Belloli EA, Beckford R, Hadley R, Flaherty KR (2016) Idiopathic non-specific interstitial pneumonia. Respirology 21: 259-268.
- Fischer A, Antoniou KM, Brown KK, Cadranel J, Corte TJ, et al. (2015) An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 46: 976-987.
- Kinder BW, Collard HR, Koth L, Daikh DI, Wolters PJ, et al. (2007) Idiopathic nonspecific interstitial pneumonia: Lung manifestation of undifferentiated connective tissue disease? American Journal of Respiratory and Critical Care Medicine 176: 691-697.
- Habiel DM, Hogaboam CM (2017) Heterogeneity of fibroblasts and myofibroblasts in pulmonary fibrosis. Current Pathobiology Reports 5: 101-110.
- Hashisako M, Fukuoka J (2015) Pathology of idiopathic interstitial pneumonias. Clinical Medicine Insights Circulatory 9: 123-133.
- Giangreco A, Reynolds SD, Stripp BR (2002) Terminal bronchioles harbor a unique airway stem cell population that localizes to the bronchoalveolar duct junction. The American Journal of Pathology 161: 173-182.
- Duffield JS, Lupher M, Thannickal VJ, Wynn TA (2013) Host responses in tissue repair and fibrosis. Annual Review of Pathology 8: 241-276.
- Friedman SL, Sheppard D, Duffield JS, Violette S (2013) Therapy for fibrotic diseases: Nearing the starting line. Science Translational Medicine 5: 167.
- Beers MF, Morrisey EE (2011) The three R’s of lung health and disease: Repair, remodeling, and regeneration. The Journal of Clinical Investigation 121: 2065-2073.
- Boström H, Willetts K, Pekny M, Levéen P, Lindahl P, et al. (1996) PDGF-A signaling is a critical event in lung alveolar myofibroblast development and alveogenesis. Cell 85: 863-873.
- Centers for Disease Control and Prevention (2020) How tobacco smoke causes disease: The biology and behavioral basis for smoking-attributable disease. Centers for Disease Control and Prevention, USA.
Citation: Tousheed SZ, Muralimohan BV (2020) Immune Response in Various Diffuse Parenchymal Lung Disorders (DPLDs). J Pulm Med Respir Res 6: 046.
Copyright: © 2020 Syed Zulkharnain Tousheed, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.