Neuroadaptations of the 5-HT System Induced by Antidepressant Treatments: Old and New Strategies
*Corresponding Author(s):
Nasser HaddjeriStem Cell And Brain Institute, INSERM U846, University Of Claude Bernard Lyon I, France
Tel:33426688215,
Fax:33478777209
Email:nasser.haddjeri@inserm.fr
Abstract
Major Depressive Disorder (MDD) is a common illness worldwide with severe socioeconomic consequences. Several hypotheses have been formulated to explain the physiopathology of depression as well as the mechanism of action of antidepressants but two of them had attracted much attention. The dominant monoaminergic hypothesis of depression links the physiopathology of MDD to a deficiency on cerebral serotonin (5-HT) and/or Norepinephrine (NE) levels; however, the relatively new neurogenic and neurotrophic hypothesis raises the possibility of an impaired neuroplasticity and/or depletion of neurotrophic factors in specific networks resulting on their structural deformity and functional impairment. An enormous body of evidence reported particular neuroadaptations following chronic administration of antidepressant drugs. In this review, we describe major adaptive changes in pre- and post-synaptic 5-HT neurotransmission as well as alterations in gene transcription and neurotrophic factors in response to long-term treatment with conventional antidepressants, new putative ones or novel promising drug candidates, all acting via 5-HT system.
Keywords
INTRODUCTION
Major Depressive Disorder (MDD) is a common illness worldwide and has a profound impact on public health. According to the Diagnostic and Statistical Manual (DSM IV-TR, [1]) of Mental Disorders, MDD is described as a depressed mood or loss of interest or pleasure (anhedonia) for more than two weeks. In addition, individuals experiencing MDD exhibit five or more symptoms (among nine possible symptoms listed in the DSM IV-TR definition) that cause significant impairment at the social, occupational and educational levels. These symptoms may include weight loss or gain, low energy, insomnia or oversleeping, psychomotor agitation or retardation, low self-esteem or suicidal ideation. A recent study estimates the prevalence of MDD at 4.4% to 5% in the general population with an annual incidence of 2.4% to 3.8% [2]. In this context, nearly one in four women and one in six men experience depression during lifetime [3]. The 12-month prevalence of MDD ranges from 2.2% in Japan to 10.45% in Brazil with similar averages of 5.5% in developed and 5.9% in developing countries [4]. The most contemporary theory of depression is the notion that stress can initiate cognitive and possibly biological processes that increase risks for the disorder. In fact, major stressful life events are one of the best predictors of an impending onset of depression and include childhood abuse [5], childhood low socioeconomic status [6], workplace mobbing [7], poor working conditions [8], loss (e.g. death of a close person) [9], humiliation (especially partner-initiated separation) [9], concomitant pathologies such as traumatic injuries [10] and cancer [11], and other stress sources. Significant socioeconomic consequences of MDD have been noted in the form of substantial loss in life quality [12], absenteeism and poor work performance [13-15], higher pharmaceutical costs [16], higher suicide risks and mortality [17-18]. Due to the high prevalence of depressive disorder, the antidepressant drugs have become one of the most common medications in western countries, with as an example of 6% of users in France [19]. However, only a small proportion of MDD patients receive adequate treatment, with only one third reported a complete remission after trial of a first antidepressant followed by progressively lower response rates with each subsequent antidepressant trial [20]. Even among responders, there is a time lag of several weeks to months before the meaningful clinical response can be observed. These drawbacks are largely due to the absence of diversity in the mode of action of conventional antidepressants, which act principally via an enhancement of synaptic transmission of the monoamines serotonin (5-HT) and/or Norepinephrine (NE) [21]. The development of this type of medications was indeed largely based on the monoaminergic hypothesis of depression that links the physiopathology of depression to a deficiency on cerebral 5-HT and/or NE levels. The first generation of ADs, Monoamine Oxidase Inhibitors (MAOIs) and Tricyclic Antidepressants (TCAs) inhibit the breakdown of 5-HT, NE and dopamine in presynaptic neurons and block the presynaptic uptake of 5-HT and NE through high-affinity 5-HT (SERT) or NE (NET) transporters, respectively. The severe side effects and toxicity of these drugs urge the development of novel drugs with a better profile, including Selective 5-HT Reuptake Inhibitors (SSRIs), NE Reuptake Inhibitors (NRIs) and combined-action 5-HT/NE reuptake inhibitors (SNRIs). However, this new generation of antidepressants acts through the modulation of monoamine transporters as the older one, which may explain their sub-optimal therapeutic efficacy. A number of emerging ADs that target monoamine transmission attempt to act on existing targets in more synergic ways (combining 5-HT reuptake inhibition with inhibition of autoreceptors) or to broaden the spectrum of monoamine systems targeted (dopamine, melatonin) to either enhance efficacy or speed response.Despite the emergence of alternative hypotheses, the monoaminergic one still dominates the research of the physiopathology and pharmacotherapy of MDD. It does not only propose the crucial involvement of monoamines in the therapeutic effects of antidepressant drugs but also suggests that depression is directly related to decreased monoaminergic transmission. The considerable potential of this hypothesis has been reinforced by the discovery of many adaptive changes, especially in the sensitivity of monoamine receptors, occurring after chronic antidepressant treatments in a time course consistent with their therapeutic action. Nevertheless, recent emphasis on neuronal circuits catalyzed a shift in our comprehension of MDD physiopathology. In particular, neurogenic and neurotrophic hypothesis of depression raises the possibility of an impaired ability of genes in specific networks, especially the hippocampus, to encode environmental stimuli and stressful events. This leads to a reduced neuroplasticity and/or depletion of neurotrophic factors, resulting in a structural deformity and functional impairment of the central nervous system.
In order to better understand the biological basis of antidepressant treatments, especially those that act through the 5-HT system, this review summarizes some of the most documented neuroadaptations, in respect to both the monoaminergic and neurogenic/neurotrophic hypothesis of depression. Attention will be paid to the neuroadaptive consequences of combination strategies (e.g. adjunction of antipsychotics), promising targets on AD development (5-HT7 receptor antagonism, 5-HT4 agonism) as well as new multimodal antidepressants.
NEUROADAPTATIONS OF THE 5-HT SYSTEM
Effects of MAOIs and TCAs
Early preclinical studies showed that acute administration of MAOIs (pargyline, tranylcypromine, phenelzine and iproniazid) and TCAs (clomipramine, imipramine, amitriptyline and nortriptyline) suppresses the firing activity of 5-HT neurons in the Dorsal Raphe Nucleus (DRN) [24-26], which can be reversed by an administration of the 5-HT1A receptors antagonist, WAY-100635 [26,27]. After long-term treatment with MAOIs, a complete recovery of the firing activity of DRN 5-HT neurons is obtained [28]. This is a consequence of a desensitization of the somatodendritic 5-HT1A autoreceptors as the agonist 8-OH-DPAT failed to affect the DRN 5-HT firing activity [28,29]. Similarly, a desensitization of these 5-HT1A autoreceptors was observed in MAO-A mutant mice [30]. Interestingly, such desensitization was attributed to an altered receptor-G protein interaction rather than a simple down-regulation, since the agonist-stimulated [35S]-GTP?S binding in the dorsal raphe was shown to be reduced after chronic administration of MAOIs such as clorgyline [31]. In contrast, chronic MAOIs do not desensitize terminal 5-HT autoreceptors. In this context, it has been shown that long-term administration of the MAOI MDL72394 had no effect on the number of 5-HT1B binding sites or the ability of the 5-HT1B agonist RU24969 to reduce extracellular levels of 5-HT in the rat frontal cortex [32,33]. At the postsynaptic level, it has been demonstrated that the selective and potent 5-HT1A antagonist WAY-100635 significantly increased the firing of CA3 pyramidal neurons in the hippocampus of rats chronically treated with the MAO-A inhibitor befloxatone, indicating an enhancement of the tonic activation of postsynaptic 5-HT1A receptors [34]. This was associated with an unaffected autoradiographic labeling of these postsynaptic receptors after repeated treatments with clorgyline [31]. Taken together, these adaptive changes, obtained after a chronic MAOI administration, are associated with the enhancement of the 5-HT neurotransmission in the brain (Figure 1) [31-34]. This data was supported by the enhanced extracellular 5-HT amounts in the ventral hippocampus, frontal cortex and DRN found in MAO-A knock-out mice compared to wild-type littermates [30].
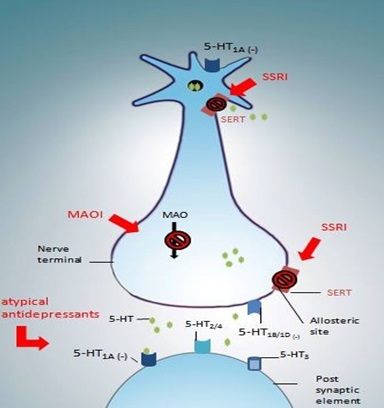 Figure 1: Mechanism of action of current antidepressant drugs.
Figure 1: Mechanism of action of current antidepressant drugs.As shown with MAOIs, long-term administration of TCAs induced a recovery of the mean firing rate of the 5-HT neurons in the DRN [29]. In contrast, an intravenous injection of the 5-HT agonist LSD or microiontophoretic application of 5-HT or LSD still decreased this firing, suggesting that the sensitivity of 5-HT autoreceptors is not modified [29]. In this context, no changes were observed in i) 5-HT1A density or mRNA expression in the DRN [35,36] or in ii) 5-HT1A agonist-stimulated [35S]-GTP?S binding in dorsal and median raphe nuclei [37,38]. These latter data suggest that neither down regulation nor desensitization of somatodendritic 5-HT1A autoreceptors occurred after chronic TCA treatments. Similarly, the sensitivity of terminal 5-HT1B/1D autoreceptors seem to be unaffected since subcutaneous injection of the 5-HT1B/1D antagonist GR127935 leads to an increase of 5-HT levels in the hippocampus and frontal cortex of rats chronically treated with the TCA clomipramine [39]. Initial observations reported that long-term administration of TCAs sensitized postsynaptic 5-HT receptors responsiveness in forebrain structures including hippocampus, amygdala, lateral geniculate nucleus and cerebral cortex [40-43]. Subsequently, long-term application of imipramine was shown to enhance the responsiveness of postsynaptic CA3 hippocampus pyramidal neurons to the microiontophoretic application of 5-HT or 8-OH-DPAT [44]. In support, chronic administration of amitriptyline increased 5-HT1A agonist-stimulated [35S]-GTP?S binding in the hippocampus without affecting the binding of [3H]8-OH-DPAT [38], indicating a hypersensitivity of postsynaptic 5-HT1A receptors non-associated with a change in the number of binding sites. It is noteworthy that no effect was observed on 5-HT1A, 5-HT2A, 5-HT2C or 5-HT7 mRNA expression in any hippocampal sub region [36]. In summary, as MAOIs treatments, TCAs induce a net increase of 5-HT neurotransmission in the brain.
Effects of SSRIs
It is extensively reported that acute administration of SSRIs produces a strong and transient increase of extracellular 5-HT levels, thus activating somatodendritic 5-HT1A autoreceptors which reduce the firing activity of DRN 5-HT neurons [46-49]. In this context, an elegant immunoelectron microscopy study demonstrated that a single injection of the SSRI fluoxetine in rats induces a decrease in the 5-HT1A immunogold labeling of the plasma membrane of the DRN dendrites and a concomitant increase in their cytoplasmic labeling, without any change in hippocampal dendrites [50]. In agreement, a reduction of the in vivo binding of the 5-HT1A Positron Emission Tomography (PET) radioligand [18F]MPPF in the DRN, but not in the hippocampus, was observed one hour after fluoxetine injection [50]. Taking together, these data indicate an internalization of the somatodendritic 5-HT1A autoreceptors. Importantly, similar autoreceptor sequestration was reported in the DRN of human volunteers after taking a single dose of fluoxetine [51]. Effects of a subchronic treatment with SSRIs on the 5-HT firing activity did not differ from those of an acute administration [52-53]. However, chronic treatments with these drugs produced a recovery of the 5-HT firing activity accompanied with a desensitization of the body 5-HT autoreceptors as shown for example by a decrease in the action of 8-OH-DPAT to reduce 5-HT levels [54-56]. Surprisingly, it has been reported that neither the number of 5-HT1A receptor-labeled dendrites in the DRN nor the density of somatodendritic 5-HT1A receptor labeling on the plasma membrane of these dendrites was significantly different from control after SSRI (fluoxetine) treatment [57]. This is in keeping with the lack of changes in the in vivo binding of [18F]MPPF in the DRN and hippocampus of rats [57] as well as in the [18F]MPPF binding potential in any cat brain region after chronic treatment with fluoxetine [58]. These data suggest the presence at the plasma membrane of DRN 5-HT neurons of uncoupled 5-HT1A autoreceptors from their G-proteins (inactivated state). In fact, it is widely admitted that after activation, many G-protein coupled receptors internalize and recycle back to the membrane as fully competent receptors. However, after chronic treatment, the repeated activation and internalization of the pool of functional 5-HT1A autoreceptors apparently led to their replacement by a pool of inactivated receptors that recycle to the plasma membrane of DRN 5-HT neurons. In this context, it has been shown that after chronic fluoxetine, the 5-HT1A agonist-stimulated [35S]-GTP?S binding was reduced [31,37,59], which is surprisingly not the case after chronic sertraline or citalopram [60,61]. This indicates that this class of antidepressants produces a differential functional regulation of 5-HT1A autoreceptors. Desensitization of terminal 5-HT1B/1D receptors with no change on their binding sites was also reported after chronic SSRI treatment [62,63]. Repeated administration of fluoxetine produced a desensitization of these terminal autoreceptors in both hippocampus and frontal cortex, but not in the hypothalamus [63,64], suggesting that the regulation of extracellular levels of 5-HT may be subject to different autoregulatory mechanism depending on the brain region. At the postsynaptic level, 5-HT1A heteroreceptors are known not to desensitize [34,62], where they are coupled to Gao protein [65]. For instance, electrophysiological recordings in brain slices from rats chronically treated with fluoxetine showed no change of the 5-HT1A-evoked responses of CA1 pyramidal cells in the hippocampus [62]. However, after long-term treatment with paroxetine, the selective 5-HT1A receptor antagonist WAY100635 markedly increased the firing activity of CA3 pyramidal neurons, a disinhibition not shown in control rats or in those treated with non-antidepressant drugs [34,66]. This indicates that SSRIs (as well as MAOIs and TCAs) might alleviate depression by enhancing the tonic activation of forebrain postsynaptic 5-HT1A receptors [34].
SERT, the site of action of SSRIs, similarly undergoes adaptive changes during SSRI treatment. Increases [67], decreases [68-70] or no change [71,72] in density of SERT binding sites have been reported in the DRN and various territories of 5-HT innervations. However, an elegant study using immunoelectron microscopy in rats indicates that there is an internalization of SERT at cell bodies and dendrites of DRN neurons as well as at their axon terminals in the hippocampus after chronic, but not acute, treatment with fluoxetine [51]. Furthermore, a reduction in the overall density of labeling in both DRN and hippocampal terminals was observed, suggesting that SERT was not only internalized, but also degraded, in the course of the chronic fluoxetine treatment [51]. This is in agreement with earlier studies showing a desensitization and a downregulation of SERT following repeated administrations of paroxetine or sertraline [68,69]. Other neuroadaptive changes were reported after chronic fluoxetine such as a desensitization of 5-HT7 receptors [53] and a downregulation of their binding sites in the hypothalamus [73], as well as a downregulation of 5-HT4 binding sites in the CA1 field of the hippocampus [74].
Effects of novel strategies
In order to avoid usual molecular targets of conventional antidepressants, many promising candidates have been identified to develop fast-acting and more effective antidepressants, including 5-HT4 receptors and 5-HT7 receptors [53,83]. It has been previously shown that a subchronic treatment (3 days) with the 5-HT4 agonist RS67333 induced a desensitization of somatodendritic 5-HT1A autoreceptors [83], increased tonic activation of hippocampal postsynaptic 5-HT1A receptors [83], increased basal 5-HT levels and decreased its metabolite 5-HIAA levels in the rat ventral hippocampus [84]. Furthermore, co-administration of the SSRI citalopram and 5-HT4 receptor agonists RS6733 or prucalopride for only 3 days exhibited similar neuroadaptive changes of the 5-HT system than those obtained after 2 to 3 weeks of citalopram application, except that the tonus of the hippocampal 5-HT1A heteroreceptors was two to three times higher [85]. This clearly indicates an important increase of the 5-HT neurotransmission following adjunctive SSRI/5-HT4 agonist treatment.
A new trend in pharmacotherapy research is the multi-target approach that may prove successful in order to find new and more effective therapies for the complexity of MDD. In this category, vilazodone, a 5-HT1A receptor partial agonist and SSRI [86-88], offers a new therapeutic possibility [89,90]. It has been shown that vilazodone induces an elevation of 5-HT amounts in the medial and the lateral cortex that is six-fold higher than those induced by SSRIs paroxetine, citalopram or fluoxetine [86]. Using in vivoelectrophysiology in the DRN of anesthetized rats, the sensitivity of 5-HT1A autoreceptors of 5-HT neurons was tested by determining the intravenous dose of the agonist 8-OH-DPAT required to suppress the basal firing rate of these neurons by 50% (ID50) after acute or subchronic (3 days) treatment with vilazodone [91]. While paroxetine or fluoxetine did not affect the ID50 value of 8-OH-DPAT, acute and subchronic administrations of vilazodone significantly increased this value, even after 24 hours in the case of a subchronic treatment [91]. This result suggests that this antidepressant induces a rapid but also prolonged inhibition of 5-HT1A autoreceptors, which can occur by either direct interaction with these receptors or their desensitization [91]. Undoubtedly, further studies are needed to determine other effects of this new antidepressant on the 5-HT system. Another multimodal-acting antidepressant is vortioxetine which acts as a 5-HT3, 5-HT7 and 5-HT1D receptor antagonist, 5-HT1B receptor partial agonist, 5-HT1A receptor agonist and SERT inhibitor [92-94]. Strikingly, 1-day treatment with vortioxetine is sufficient to induce a recovery of the 5-HT neuronal firing activity in the DRN, an effect observed after 14 days of fluoxetine treatment [93]. This markedly faster recovery was associated with 5-HT3 receptor antagonism and reduced SERT occupancy [93]. Additional data showed that acute or subchronic 3-day administration of vortioxetine caused robust dose-dependent increase of extracellular 5-HT levels in the ventral hippocampus and the median PFC, with a greater effect on the former compared to the latter [92]. This increase seems to occur at low levels of SERT occupancy. Further research is required to establish the full effects of the unique pharmacological and neurochemical profile of this antidepressant.
Taken together, these data indicates that a change in the 5-HT receptor sensitivity at the presynaptic and/or postsynaptic level can be a common mechanism of action of the 5-HT antidepressants. These neuroadaptations took place in a time course consistent with the onset of action of each antidepressant strategy. Such neuroadaptive changes are a major argument supporting the monoaminergic hypothesis. However, a growing body of evidence also shows that downstream antidepressant actions include adaptations of signal transduction pathways and gene expression, notably neurotrophic factors and hippocampal neurogenesis [95-97], which favour the neurotrophic and neurogenic hypothesis of depression. In the following part of this review, we will describe the neuroadaptive changes seen after 5-HT antidepressant treatment according to this theory.
NEUROTROPHIC AND NEUROGENIC ADAPTATIONS INDUCED BY 5-HT ANTIDEPRESSANTS
Neurotrophins
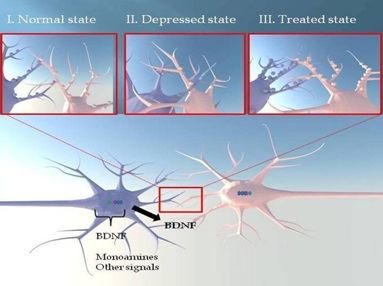
Antidepressants could oppose or reverse the action of stress on the 5-HT system via a positive effect on cerebral BDNF. Indeed, it has been reported that chronic treatment with different classes of antidepressants (SSRIs fluoxetine and escitalopram, MAOIs tranylcypromine and phenelzine, TCA desipramine) increases BDNF brain levels [101,110-113]. A very recent study reported that long-term administration of the SSRI escitalopram to mice, experiencing postnatal maternal separation plus 4 weeks of UCMS in adolescence, induced an elevation of BDNF levels in the hippocampus [114]. This result was obtained when stressed mice were treated either during or following exposure to stress, demonstrating that antidepressants are capable of blocking and reversing the negative effect of stress via BDNF actions [114]. Such modulation of BDNF concentrations seemed to be time-dependent. Indeed, a previous study monitoring BDNF mRNA in the rat hippocampus reported that daily oral administration of fluoxetine decreased this expression after 4 days, had no effect after 7 days and enhanced it after 14 days [110]. This biphasic response can be explained by a differential transcript regulation since rat BDNF gene expresses four mRNA isoforms that are modulated by different signaling cascades. In this context, acute injection of fluoxetine or tranylcypromine was reported to decrease exon V and IV mRNAs without affecting those of exon I or III, whereas prolonged treatment enhanced expression of exon V and exon I mRNAs without changing those of exon III or IV [115]. Clinical data are in accordance with preclinical results and show an increase of serum or platelet BDNF levels following several weeks of antidepressant treatment. For example, depressed patients showed an elevated plasma BDNF level after chronic treatment with SSRI paroxetine or citalopram compared to drug-free patients [104,116]. Interestingly, a recent pilot study showed that plasma BDNF of depressed patients increased rapidly (within 7 days) after the initiation of an antidepressant (including SSRIs and TCAs) treatment, and in turn, the absence of this early increase seemed to indicate that the selected treatment will not be effective [117]. This suggests a valuable clinical utility of peripheral BDNF measurement as surrogate marker of antidepressant efficacy [117].
There is a lack of data describing the effects of novel antidepressants targeting the 5-HT system on BDNF. Previous work from Li's lab [118] demonstrated that chronic restraint stress reduced BDNF expression in the rat hippocampus and that chronic administration of quetiapine or venlafaxine dose-dependently prevented this reduction. Interestingly, combination of lower doses of quetiapine and venlafaxine prevented BDNF decrease in stressed rats, whereas each of the drugs exerted mild or no effects [118]. These data suggest that both drugs share the hippocampus as their common target by enhancing hippocampal resilience, which may be impaired in MDD patients. In accordance, a clinical study assessing the efficacy of risperidone addition on sertraline-resistant patients showed that, 4 weeks after risperidone co-treatment, plasma BDNF levels were increased in responders (reduced HAM-D scores), whereas no change was observed in non-responders [119]. This supports the idea that enhanced levels of peripheral BDNF can represent a biological marker of the antidepressant response. Another study investigating the effects of a new promising target reported an enhancement of BDNF protein levels in the hippocampus of rats after only a subchronic application of the 5-HT4 receptor agonist SL65.0155, which was not obtained using subchronic citalopram or clomipramine treatments [120], again in support of the fast-acting antidepressant profile of 5-HT4 receptor agonists.
Numerous antidepressant treatments have been shown to affect VEGF signaling. Elegant studies from Duman's group reported that chronic treatment with SSRIs fluoxetine or sertraline, TCAs desipramine or amitriptyline and SNRI venlafaxine increases VEGF protein levels in the hippocampus [132,133]. Interestingly, such regulation seems to require the activation of the 5-HT1A receptor subtype, at least, in the case of fluoxetine [133]. However, data reported in cultured cell systems are not consistent. For example, a treatment with the SSRIs fluoxetine or paroxetine in cultured cortical astrocytes leads to an up regulation of the VEGF mRNA levels whereas the TCAs imipramine and desipramine did not affect this expression [134]. The latter result suggests that SSRIs may contribute to normalize the trophic support to neurons in major depression by increasing the expression of this specific astrocyte-derived neurotrophic factor, while TCAs may involve other astrocytic mechanisms or other cell populations to accomplish their therapeutic effects [134]. Interestingly, it has been reported that, after a sub-acute treatment in rats, neither the SSRI citalopram nor TCA clomipramine affected the VEGF protein levels in hippocampal homogenates, unlike the 5-HT4 receptor agonist SL65.0155 which increased these levels, accordingly to its fast-acting antidepressant profile [120]. The clinical data shed light to a more complex landscape. An eight-week treatment with paroxetine appeared to decrease VEGF mRNA levels in peripheral leukocytes of depressed patients in positive correlation with clinical improvement [125]. In the same class of antidepressants, escitalopram did not alter VEGF plasma levels in depressed patients after 12 weeks of treatment [129]. It has to be mentioned that this latter study was conducted in a very small sample (25 subjects) without control of the co-morbidities, cardiovascular diseases or confounding factors, making conclusions very hard to make [129].
It is of high interest to note that these neurotrophic factors and related signaling pathways are associated with antidepressant action and could operate as key modulators in the regulation of neurogenesis in the adult hippocampus [135]. The findings that antidepressants increase neurotrophic factor expression, particularly in the hippocampus, provide the background and the rationale for studies of adult neurogenesis and its implication on the etiology of depression and the antidepressant response.
One of the most reproducible findings in antidepressant research is that different classes of antidepressants including TCAs, MAOIs and SSRIs increase hippocampal cell proliferation and neurogenesis [83, 139,144-146] after chronic, but not acute, treatment consistent with the time course for their onset of therapeutic action. Similar positive effects have been reported using novel therapeutic strategies. For example, quetiapine add-on therapy increased the number of newborn cells in the hippocampus of fluoxetine treatment-resistant rats that experienced UCMS [147]. Putative fast-acting antidepressants, including 5-HT7 receptor antagonist SB-269970 [53] and 5-HT4 receptor agonists RS67333 and prucalopride [83,85] induced an enhancement of hippocampal neurogenesis in a time course concordant with their onset of action. Very recently, a preclinical study using the multimodal antidepressant vortioxetine showed that a 21-day treatment with this drug was able to increase cell proliferation and survival and to stimulate the maturation of immature granule cells in the SGZ of the DG of the hippocampus [146] while a preliminary study revealed that such DG cell proliferation increase occurred after one day of treatment only [148]. Taken together, these data indicate that up regulation of hippocampal neurogenesis may be a common dominator of the mechanism of action of antidepressants. However, the function of these newly formed neurons is still not fully understood. Previous studies suggested that young granule cells constitute a distinct population that exhibit a greater degree of plasticity than mature neurons: i) they displayed a reduced threshold to induction of Long-Term Potentiation (LTP) [149], and ii) they can be tonically activated by ambient GABA before being sequentially innervated by GABA- and glutamate-mediated synaptic inputs, leading to marked defects in their synapse formation and dendritic development in vivo [150]. This hypothesis has to be confirmed by additional studies.
CONCLUSION
The list of neuroadaptations induced by 5-HT antidepressants mentioned in this review is certainly not exhaustive. Establishing the full profile of these neuroadaptive changes is necessary to better target the downstream common mechanisms. Although the monoamine imbalance is undoubtedly involved, the classical monoaminergic systems seemed to be simplistic over the years. The only elevation of monoamine neurotransmissions could not explain the temporal delay in the therapeutic action of antidepressants. In the same way, the neurogenic/neurotrophic hypothesis could not support the physiopathology of MDD or the antidepressant response by only the enhancement of neurotrophin expression and/or addition of new cells to existing relevant networks. These two hypotheses can be considered complementary as activation of monoamine receptors may modulate the expression of intracellular proteins and growth factors, but still not sufficient to explain the heterogeneity of MDD symptoms and the failure of antidepressant effect in some cases. The elaboration of a putative more efficient model has to be addressed. Other hypotheses on MDD pathophysiology are currently under investigation, such as possible alterations of the glial system, or genetic and epigenetic modifications. The modulatory role of each candidate is still far from being understood. Future works will definitely come up with a new way to approach MDD in order to understand its etiology and to develop new therapeutics with broader efficacy.
Depression is associated with reduced levels of monoamines in the brain, such as 5-HT. Chronic administration of antidepressants can cause a number of changes in the brain, depending on the particular drug type. Selective 5-HT Reuptake Inhibitors (SSRIs) and Monoamine Oxidase (MAO) Inhibitors (MAOIs) enhance 5-HT extracellular levels by blocking the 5-HT transporter (SERT) or by inhibiting the 5-HT degradation, respectively. Chronic administration of these drugs desensitizes somatodendritic 5-HT1A receptors and terminal 5-HT1B/1D autoreceptors leading to an enhancement of 5-HT neurotransmission.
There is a possible contribution of neuronal plasticity in the action of SERT ligands.
I/ A normal hippocampal pyramidal neuron is represented. This neuron is regulated by Brain-Derived Neurotrophic Factor (BDNF) and can be innervated by monoaminergic and other signals.
II/ In depressed state, there is a decrease of brain levels of monoamines and neurotrophic factors inducing atrophy of dendritic arborization.
III/ Antidepressants might reverse the effects of stress. This can be observed by an increase in the length and density of dendritic extensions, promoting the formation of synaptic contacts. These effects could explain the onset of antidepressant response obtained with this type of SERT-targeting antidepressants.
REFERENCES
- Lawson AK, Wright CV, Fitzgerald LF (2013) The evaluation of sexual harassment litigants: reducing discrepancies in the diagnosis of posttraumatic stress disorder. Law Hum Behav 37: 337-347.
- Ferrari AJ, Somerville AJ, Baxter AJ, Norman R, Patten SB, et al. (2013) Global variation in the prevalence and incidence of major depressive disorder: a systematic review of the epidemiological literature. Psychol Med 43: 471-481.
- Kessler RC, Birnbaum H, Bromet E, Hwang I, Sampson N, et al. (2010) Age differences in major depression: results from the National Comorbidity Survey Replication (NCS-R). Psychol Med 40: 225-237.
- Bromet E, Andrade LH, Hwang I, Sampson NA, Alonso J, et al. (2011) Cross-national epidemiology of DSM-IV major depressive episode. BMC Med 9: 90.
- Comijs HC, van Exel E, van der Mast RC, Paauw A, Oude Voshaar R, et al. (2013) Childhood abuse in late-life depression. J Affect Disord 147: 241-246.
- Ochi M, Fujiwara T, Mizuki R, Kawakami N (2014) Association of socioeconomic status in childhood with major depression and generalized anxiety disorder: results from the World Mental Health Japan survey 2002-2006. BMC public health 14: 359.
- Kostev K, Rex J, Waehlert L, Hog D, Heilmaier C (2014) Risk of psychiatric and neurological diseases in patients with workplace mobbing experience in Germany: a retrospective database analysis. Ger Med Sci 12: 10.
- Plaisier I, de Bruijn JG, de Graaf R, ten Have M, Beekman AT, et al. (2007) The contribution of working conditions and social support to the onset of depressive and anxiety disorders among male and female employees. Soc Sci Med 64: 401-410.
- Kendler KS, Hettema JM, Butera F, Gardner CO, Prescott CA (2003) Life event dimensions of loss, humiliation, entrapment, and danger in the prediction of onsets of major depression and generalized anxiety. Arch Gen Psychiatry 60: 789-796.
- Warren AM, Foreman ML, Bennett MM, Petrey LB, Reynolds M, et al. (2014) Posttraumatic stress disorder following traumatic injury at 6 months: associations with alcohol use and depression. J Trauma Acute Care Surg 76: 517-522.
- Mitchell AJ, Chan M, Bhatti H, Halton M, Grassi L, et al. (2011) Prevalence of depression, anxiety, and adjustment disorder in oncological, haematological, and palliative-care settings: a meta-analysis of 94 interview-based studies. Lancet Oncol 12: 160-174.
- de Graaf R, Bijl RV, Smit F, Vollebergh WA, Spijker J (2002) Risk factors for 12-month comorbidity of mood, anxiety, and substance use disorders: findings from the Netherlands Mental Health Survey and Incidence Study. Am J Psychiatry 159: 620-629.
- Plaisier I, de Graaf R, de Bruijn J, Smit J, van Dyck R, et al. (2012) Depressive and anxiety disorders on-the-job: the importance of job characteristics for good work functioning in persons with depressive and anxiety disorders. Psychiatry Res 200: 382-388.
- Bertilsson M, Petersson EL, Ostlund G, Waern M, Hensing G (2013) Capacity to work while depressed and anxious--a phenomenological study. Disabil Rehabil 35: 1705-1711.
- Wada K, Arakida M, Watanabe R, Negishi M, Sato J, et al. (2013) The economic impact of loss of performance due to absenteeism and presenteeism caused by depressive symptoms and comorbid health conditions among Japanese workers. Ind Health 51: 482-489.
- Bock JO, Luppa M, Brettschneider C, Riedel-Heller S, Bickel H, et al. (2014) Impact of depression on health care utilization and costs among multimorbid patients--from the MultiCare Cohort Study. PLoS One 9: 91973.
- Olin B, Jayewardene AK, Bunker M, Moreno F (2012) Mortality and suicide risk in treatment-resistant depression: an observational study of the long-term impact of intervention. PLoS One 7: 48002.
- Finlay AK, Oliva EM, Timko C, Moos RH, Cronkite R (2014) Predictors of 30-year mortality in depressed and comparison samples. J Affect Disord 165: 114-119.
- Gasquet I, Gury C, Tcherny-Lessenot S, Quesnot A, Gaudebout P (2005) Patterns of prescription of four major antipsychotics: a retrospective study based on medical records of psychiatric inpatients. Pharmacoepidemiol Drug Saf 14: 805-811.
- Fabbri C, Marsano A, Balestri M, De Ronchi D, Serretti A (2013) Clinical features and drug induced side effects in early versus late antidepressant responders. J Psychiatr Res 47: 1309-1318.
- Jayanthi LD, Ramamoorthy S (2005) Regulation of monoamine transporters: influence of psychostimulants and therapeutic antidepressants. AAPS J 7: 728-738.
- Miller J (2004) Managing antidepression overdoses. Emerg Med Serv 33: 113-119.
- Grady MM, Stahl SM (2012) Practical guide for prescribing MAOIs: debunking myths and removing barriers. CNS Spectr 17: 2-10.
- Aghajanian GK, Graham AW, Sheard MH (1970) Serotonin-containing neurons in brain: depression of firing by monoamine oxidase inhibitors. Science 169: 1100-1102.
- Scuvée-Moreau JJ, Dresse AE (1979) Effect of various antidepressant drugs on the spontaneous firing rate of locus coeruleus and dorsal raphe neurons of the rat. Eur J Pharmacol 57: 219-225.
- Gartside SE, Umbers V, Sharp T (1997) Inhibition of 5-HT cell firing in the DRN by non-selective 5-HT reuptake inhibitors: studies on the role of 5-HT1A autoreceptors and noradrenergic mechanisms. Psychopharmacology (Berl) 130: 261-268.
- Sharp T, Gartside SE, Umbers V (1997) Effects of co-administration of a monoamine oxidase inhibitor and a 5-HT1A receptor antagonist on 5-hydroxytryptamine cell firing and release. Eur J Pharmacol 320: 15-19.
- Blier P, de Montigny C (1985) Serotoninergic but not noradrenergic neurons in rat central nervous system adapt to long-term treatment with monoamine oxidase inhibitors. Neuroscience 16: 949-955.
- Blier P, de Montigny C (1980) Effect of chronic tricylic antidepressant treatment on the serotoninergic autoreceptor: a microiontophoretic study in the rat. Naunyn Schmiedebergs Arch Pharmacol 314: 123-128.
- Evrard A, Malagié I, Laporte AM, Boni C, Hanoun N, et al. (2002) Altered regulation of the 5-HT system in the brain of MAO-A knock-out mice. Eur J Neurosci 15: 841-851.
- Shen C1, Li H, Meller E (2002) Repeated treatment with antidepressants differentially alters 5-HT1A agonist-stimulated [35S]GTP gamma S binding in rat brain regions. Neuropharmacology 42: 1031-1038.
- Palfreyman MG, Mir AK, Kubina M, Middlemiss DN, Richards M, et al. (1986) Monoamine receptor sensitivity changes following chronic administration of MDL 72394, a site-directed inhibitor of monoamine oxidase. Eur J Pharmacol 130: 73-89.
- Sleight AJ, Smith RJ, Marsden CA, Palfreyman MG (1989) The effects of chronic treatment with amitriptyline and MDL 72394 on the control of 5-HT release in vivo. Neuropharmacology 28: 477-480.
- Haddjeri N, Blier P, de Montigny C (1998) Long-term antidepressant treatments result in a tonic activation of forebrain 5-HT1A receptors. J Neurosci 18: 10150-10156.
- Gozlan H, Saddiki-Traki F, Merahi N, Laguzzi R, Hamon M (1991) [Preclinical pharmacology of amoxapine and amitriptyline. Implications of serotoninergic and opiodergic systems in their central effect in rats]. Encephale 3: 415-422.
- Yau JL, Olsson T, Noble J, Seckl JR (1999) Serotonin receptor subtype gene expression in the hippocampus of aged rats following chronic amitriptyline treatment. Brain Res Mol Brain Res 70: 282-287.
- Hensler JG (2002) Differential regulation of 5-HT1A receptor-G protein interactions in brain following chronic antidepressant administration. Neuropsychopharmacology 26: 565-573.
- Rossi DV, Valdez M, Gould GG, Hensler JG (2006) Chronic administration of venlafaxine fails to attenuate 5-HT1A receptor function at the level of receptor-G protein interaction. Int J Neuropsychopharmacol 9: 393-406.
- Gur E, Lerer B, Newman ME (1999) Chronic clomipramine and triiodothyronine increase serotonin levels in rat frontal cortex in vivo: relationship to serotonin autoreceptor activity. J Pharmacol Exp Ther 288: 81-87.
- de Montigny C, Aghajanian GK (1978) Tricyclic antidepressants: long-term treatment increases responsivity of rat forebrain neurons to serotonin. Science 202: 1303-1306.
- Wang RY, Aghajanian GK (1980) Enhanced sensitivity of amygdaloid neurons to serotonin and norepinephrine after chronic antidepressant treatment. Commun Psychopharmacol 4: 83-90.
- Jones RS (1980) Long-term administration of atropine, imipramine, and viloxazine alters responsiveness of rat cortical neurones to acetylcholine. Can J Physiol Pharmacol 58: 531-535.
- Menkes DB, Aghajanian GK (1981) alpha 1-Adrenoceptor-mediated responses in the lateral geniculate nucleus are enhanced by chronic antidepressant treatment. Eur J Pharmacol 74: 27-35.
- Chaput Y, de Montigny C, Blier P (1991) Presynaptic and postsynaptic modifications of the serotonin system by long-term administration of antidepressant treatments. An in vivo electrophysiologic study in the rat. Neuropsychopharmacology 5: 219-229.
- Cryan JF, Markou A, Lucki I (2002) Assessing antidepressant activity in rodents: recent developments and future needs. Trends Pharmacol Sci 23: 238-245.
- Invernizzi R, Belli S, Samanin R (1992) Citalopram's ability to increase the extracellular concentrations of serotonin in the dorsal raphe prevents the drug's effect in the frontal cortex. Brain Res 584: 322-324.
- Bel N, Artigas F (1992) Fluvoxamine preferentially increases extracellular 5-hydroxytryptamine in the raphe nuclei: an in vivo microdialysis study. Eur J Pharmacol 229: 101-103.
- Mansari ME, Wiborg O, Mnie-Filali O, Benturquia N, Sánchez C, et al. (2007) Allosteric modulation of the effect of escitalopram, paroxetine and fluoxetine: in-vitro and in-vivo studies. Int J Neuropsychopharmacol 10: 31-40.
- Mnie-Filali O, El Mansari M, Espana A, Sànchez C, Haddjeri N (2006) Allosteric modulation of the effects of the 5-HT reuptake inhibitor escitalopram on the rat hippocampal synaptic plasticity. Neurosci Lett 395: 23-27.
- Riad M, Zimmer L, Rbah L, Watkins KC, Hamon M, et al. (2004) Acute treatment with the antidepressant fluoxetine internalizes 5-HT1A autoreceptors and reduces the in vivo binding of the PET radioligand [18F]MPPF in the nucleus raphe dorsalis of rat. J Neurosci 24: 5420-5426.
- Descarries L, Riad M (2012) Effects of the antidepressant fluoxetine on the subcellular localization of 5-HT1A receptors and SERT. Philos Trans R Soc Lond B Biol Sci 367: 2416-2425.
- Czachura JF, Rasmussen K (2000) Effects of acute and chronic administration of fluoxetine on the activity of serotonergic neurons in the dorsal raphe nucleus of the rat. Naunyn Schmiedebergs Arch Pharmacol 362: 266-275.
- Mnie-Filali O, Faure C, Lambás-Señas L, El Mansari M, Belblidia H, et al. (2011) Pharmacological blockade of 5-HT7 receptors as a putative fast acting antidepressant strategy. Neuropsychopharmacology 36: 1275-1288.
- Rutter JJ, Auerbach SB (1993) Acute uptake inhibition increases extracellular serotonin in the rat forebrain. J Pharmacol Exp Ther 265: 1319-1324.
- Kreiss DS, Lucki I (1995) Effects of acute and repeated administration of antidepressant drugs on extracellular levels of 5-hydroxytryptamine measured in vivo. J Pharmacol Exp Ther 274: 866-876.
- Dawson LA, Nguyen HQ, Smith DL, Schechter LE (2002) Effect of chronic fluoxetine and WAY-100635 treatment on serotonergic neurotransmission in the frontal cortex. J Psychopharmacol 16: 145-152.
- Riad M, Rbah L, Verdurand M, Aznavour N, Zimmer L, et al. (2008) Unchanged density of 5-HT(1A) autoreceptors on the plasma membrane of nucleus raphe dorsalis neurons in rats chronically treated with fluoxetine. Neuroscience 151: 692-700.
- Aznavour N, Rbah L, Riad M, Reilhac A, Costes N, et al. (2006) A PET imaging study of 5-HT(1A) receptors in cat brain after acute and chronic fluoxetine treatment. Neuroimage 33: 834-842.
- Castro M, Diaz A, del Olmo E, Pazos A (2003) Chronic fluoxetine induces opposite changes in G protein coupling at pre and postsynaptic 5-HT1A receptors in rat brain. Neuropharmacology 44: 93-101.
- Rossi DV, Burke TF, McCasland M, Hensler JG (2008) Serotonin-1A receptor function in the dorsal raphe nucleus following chronic administration of the selective serotonin reuptake inhibitor sertraline. J Neurochem 105: 1091-1099.
- Moulin-Sallanon M, Charnay Y, Ginovart N, Perret P, Lanfumey L, et al. (2009) Acute and chronic effects of citalopram on 5-HT1A receptor-labeling by [18F]MPPF and -coupling to receptors-G proteins. Synapse 63: 106-116.
- Le Poul E, Boni C, Hanoun N, Laporte AM, Laaris N, et al. (2000) Differential adaptation of brain 5-HT1A and 5-HT1B receptors and 5-HT transporter in rats treated chronically with fluoxetine. Neuropharmacology 39: 110-122.
- Dremencov E, Gur E, Lerer B, Newman ME (2000) Subchronic fluoxetine administration to rats: effects on 5-HT autoreceptor activity as measured by in vivo microdialysis. Eur Neuropsychopharmacol 10: 229-236.
- Newman ME, Shalom G, Ran A, Gur E, Van de Kar LD (2004) Chronic fluoxetine-induced desensitization of 5-HT1A and 5-HT1B autoreceptors: regional differences and effects of WAY-100635. Eur J Pharmacol 486: 25-30.
- Valdizán EM, Castro E, Pazos A (2010) Agonist-dependent modulation of G-protein coupling and transduction of 5-HT1A receptors in rat dorsal raphe nucleus. Int J Neuropsychopharmacol 13: 835-843.
- El Mansari M, Sánchez C, Chouvet G, Renaud B, Haddjeri N (2005) Effects of acute and long-term administration of escitalopram and citalopram on serotonin neurotransmission: an in vivo electrophysiological study in rat brain. Neuropsychopharmacology 30: 1269-1277.
- Hrdina PD, Vu TB (1993) Chronic fluoxetine treatment upregulates 5-HT uptake sites and 5-HT2 receptors in rat brain: an autoradiographic study. Synapse 14: 324-331.
- Piñeyro G, Blier P, Dennis T, de Montigny C (1994) Desensitization of the neuronal 5-HT carrier following its long-term blockade. J Neurosci 14: 3036-3047.
- Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, et al. (1999) Effects of chronic antidepressant treatments on serotonin transporter function, density, and mRNA level. J Neurosci 19: 10494-10501.
- Benmansour S, Owens WA, Cecchi M, Morilak DA, Frazer A (2002) Serotonin clearance in vivo is altered to a greater extent by antidepressant-induced downregulation of the serotonin transporter than by acute blockade of this transporter. J Neurosci 22: 6766-6772.
- Cheetham SC, Viggers JA, Slater NA, Heal DJ, Buckett WR (1993) [3H]paroxetine binding in rat frontal cortex strongly correlates with [3H]5-HT uptake: effect of administration of various antidepressant treatments. Neuropharmacology 32: 737-743.
- Spurlock G, Buckland P, O'Donovan M, McGuffin P (1994) Lack of effect of antidepressant drugs on the levels of mRNAs encoding serotonergic receptors, synthetic enzymes and 5HT transporter. Neuropharmacology 33: 433-440.
- Sleight AJ, Carolo C, Petit N, Zwingelstein C, Bourson A (1995) Identification of 5-hydroxytryptamine7 receptor binding sites in rat hypothalamus: sensitivity to chronic antidepressant treatment. Mol Pharmacol 47: 99-103.
- Vidal R, Valdizán EM, Mostany R, Pazos A, Castro E (2009) Long-term treatment with fluoxetine induces desensitization of 5-HT4 receptor-dependent signalling and functionality in rat brain. J Neurochem 110: 1120-1127.
- Han DH, Kim SM, Choi JE, Min KJ, Renshaw PF (2013) Adjunctive aripiprazole therapy with escitalopram in patients with co-morbid major depressive disorder and alcohol dependence: clinical and neuroimaging evidence. J Psychopharmacol 27: 282-291.
- Wade RL, Kindermann SL, Hou Q, Thase ME (2014) Comparative assessment of adherence measures and resource use in SSRI/SNRI-treated patients with depression using second-generation antipsychotics or L-methylfolate as adjunctive therapy. J Manag Care Pharm 20: 76-85.
- Koenig AM, Butters MA, Begley A, Ogbagaber S, Wahed AS, et al. (2014) Response to antidepressant medications in late-life depression across the spectrum of cognitive functioning. J Clin Psychiatry 75: 100-107.
- Brunner E, Tohen M, Osuntokun O, Landry J, Thase ME (2014) Efficacy and safety ofolanzapine/fluoxetine combination vs fluoxetine monotherapy following successful combination therapy of treatment-resistant major depressive disorder. Neuropsychopharmacology doi: 10.1038/npp.2014.101.
- Chernoloz O, El Mansari M, Blier P (2012) Effects of sustained administration of quetiapine alone and in combination with a serotonin reuptake inhibitor on norepinephrine and serotonin transmission. Neuropsychopharmacology 37: 1717-1728.
- Szabo ST, Blier P (2001) Serotonin (1A) receptor ligands act on norepinephrine neuron firing through excitatory amino acid and GABA(A) receptors: a microiontophoretic study in the rat locus coeruleus. Synapse 42: 203-212.
- Dremencov E, El Mansari M, Blier P (2007) Distinct electrophysiological effects of paliperidone and risperidone on the firing activity of rat serotonin and norepinephrine neurons. Psychopharmacology (Berl) 194: 63-72.
- Marcus MM, Jardemark K, Malmerfelt A, Gertow J, Konradsson-Geuken A, et al. (2012) Augmentation by escitalopram, but not citalopram or R-citalopram, of the effects of low-dose risperidone: behavioral, biochemical, and electrophysiological evidence. Synapse 66: 277-290.
- Lucas G, Rymar VV, Du J, Mnie-Filali O, Bisgaard C, et al. (2007) Serotonin(4) (5-HT(4)) receptor agonists are putative antidepressants with a rapid onset of action. Neuron 55: 712-725.
- Licht CL, Knudsen GM, Sharp T (2010) Effects of the 5-HT(4) receptor agonist RS67333 and paroxetine on hippocampal extracellular 5-HT levels. Neurosci Lett 476: 58-61.
- Lucas G, Du J, Romeas T, Mnie-Filali O, Haddjeri N, et al. (2010) Selective serotonin reuptake inhibitors potentiate the rapid antidepressant-like effects of serotonin4 receptor agonists in the rat. PLoS One 5: 9253.
- van Amsterdam C, Seyfried CA (2014) Mechanism of action of the bimodal antidepressant vilazodone: evidence for serotonin1A-receptor-mediated auto-augmentation of extracellular serotonin output. Psychopharmacology (Berl) 231: 2547-2558.
- Mandrioli R, Mercolini L, Saracino MA, Raggi MA (2012) Selective Serotonin Reuptake Inhibitors (SSRIs): therapeutic drug monitoring and pharmacological interactions. Curr Med Chem 19: 1846-1863.
- Reed CR, Kajdasz DK, Whalen H, Athanasiou MC, Gallipoli S, et al. (2012) The efficacy profile of vilazodone, a novel antidepressant for the treatment of major depressive disorder. Curr Med Res Opin 28: 27-39.
- Reinhold JA, Mandos LA, Lohoff FW, Rickels K (2012) Evidence for the use of vilazodone in the treatment of major depressive disorder. Expert Opin Pharmacother 13: 2215-2224.
- Jain R, Chen D, Edwards J, Mathews M (2014) Early and sustained improvement with vilazodone in adult patients with major depressive disorder: post hoc analyses of two phase III trials. Curr Med Res Opin 30: 263-270.
- Ashby CR, Kehne JH, Bartoszyk GD, Renda MJ, Athanasiou M, et al. (2013) Electrophysiological evidence for rapid 5-HT?A autoreceptor inhibition by vilazodone, a 5-HT?A receptor partial agonist and 5-HT reuptake inhibitor. Eur J Pharmacol 714: 359-365.
- Pehrson AL, Cremers T, Bétry C, van der Hart MG, Jørgensen L, et al. (2013) Lu AA21004, a novel multimodal antidepressant, produces regionally selective increases of multiple neurotransmitters--a rat microdialysis and electrophysiology study. Eur Neuropsychopharmacol 23: 133-145.
- Bétry C, Pehrson AL, Etiévant A, Ebert B, Sánchez C, et al. (2013) The rapid recovery of 5-HT cell firing induced by the antidepressant vortioxetine involves 5-HT(3) receptor antagonism. Int J Neuropsychopharmacol 16: 1115-1127.
- Fu J, Chen Y (2014) The efficacy and safety of 5 mg/d Vortioxetine compared to placebo for major depressive disorder: A meta-analysis. Psychopharmacology (Berl).
- Schmidt HD, Duman RS (2007) The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive-like behavior. Behav Pharmacol 18: 391-418.
- Sahay A, Drew MR, Hen R (2007) Dentate gyrus neurogenesis and depression. Prog Brain Res 163: 697-722.
- Neto FL, Borges G, Torres-Sanchez S, Mico JA, Berrocoso E (2011) Neurotrophins role in depression neurobiology: a review of basic and clinical evidence. Curr Neuropharmacol 9: 530-552.
- Huang EJ, Reichardt LF (2003) Trk receptors: roles in neuronal signal transduction. Annu Rev Biochem 72: 609-642.
- Duman RS, Monteggia LM (2006) A neurotrophic model for stress-related mood disorders. Biol Psychiatry 59: 1116-1127.
- Maturana MJ, Pudell C, Targa AD, Rodrigues LS, Noseda AC, et al. (2014) REM Sleep Deprivation Reverses Neurochemical and Other Depressive-Like Alterations Induced by Olfactory Bulbectomy. Mol Neurobiol.
- Doron R, Lotan D, Einat N, Yaffe R, Winer A, et al. (2014) A novel herbal treatment reduces depressive-like behaviors and increases BDNF levels in the brain of stressed mice. Life Sci 94: 151-157.
- Fischer CW, Liebenberg N, Elfving B, Lund S, Wegener G (2012) Isolation-induced behavioural changes in a genetic animal model of depression. Behav Brain Res 230: 85-91.
- Kyeremanteng C, MacKay JC, James JS, Kent P, Cayer C, et al. (2014) Effects of electroconvulsive seizures on depression-related behavior, memory and neurochemical changes in Wistar and Wistar-Kyoto rats. Prog Neuropsychopharmacol Biol Psychiatry 54: 170-178.
- Munno D, Sterpone S, Fania S, Cappellin F, Mengozzi G, et al. (2013) Plasma brain derived neurotrophic factor levels and neuropsychological aspects of depressed patients treated with paroxetine. Panminerva Med 55: 377-384.
- Li Z, Zhang C, Fan J, Yuan C, Huang J, et al. (2014) Brain-derived neurotrophic factor levels and bipolar disorder in patients in their first depressive episode: 3-year prospective longitudinal study. Br J Psychiatry 205: 29-35.
- Hong W, Fan J, Yuan C, Zhang C, Hu Y, et al. (2014) Significantly decreased mRNA levels of BDNF and MEK1 genes in treatment-resistant depression. Neuroreport 25: 753-755.
- Choi MJ, Kang RH, Lim SW, Oh KS, Lee MS (2006) Brain-derived neurotrophic factor gene polymorphism (Val66Met) and citalopram response in major depressive disorder. Brain Res 1118: 176-182.
- Kang HJ, Voleti B, Hajszan T, Rajkowska G, Stockmeier CA, et al. (2012) Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med 18: 1413-1417.
- Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, et al. (2012) Brain-derived neurotrophic factor Val66Met polymorphism and antidepressant efficacy of ketamine in depressed patients. Biol Psychiatry 72: 27-28.
- De Foubert G, Carney SL, Robinson CS, Destexhe EJ, Tomlinson R, et al. (2004) Fluoxetine-induced change in rat brain expression of brain-derived neurotrophic factor varies depending on length of treatment. Neuroscience 128: 597-604.
- Balu DT, Hoshaw BA, Malberg JE, Rosenzweig-Lipson S, Schechter LE, et al. (2008) Differential regulation of central BDNF protein levels by antidepressant and non-antidepressant drug treatments. Brain Res 1211: 37-43.
- Zhang Y, Gu F, Chen J, Dong W (2010) Chronic antidepressant administration alleviates frontal and hippocampal BDNF deficits in CUMS rat. Brain Res 1366: 141-148.
- Assareh N, ElBatsh MM, Marsden CA, Kendall DA (2012) The effects of chronic administration of tranylcypromine and rimonabant on behaviour and protein expression in brain regions of the rat. Pharmacol Biochem Behav 100: 506-512.
- Doron R, Lotan D, Versano Z, Benatav L, Franko M, et al. (2014) Escitalopram or novel herbal mixture treatments during or following exposure to stress reduce anxiety-like behavior through corticosterone and BDNF modifications. PloS one 9: 91455.
- Khundakar AA, Zetterström TS (2006) Biphasic change in BDNF gene expression following antidepressant drug treatment explained by differential transcript regulation. Brain Res 1106: 12-20.
- Haghighi M, Salehi I, Erfani P, Jahangard L, Bajoghli H, et al. (2013) Additional ECT increases BDNF-levels in patients suffering from major depressive disorders compared to patients treated with citalopram only. J Psychiatr Res 47: 908-915.
- Dreimüller N, Schlicht KF, Wagner S, Peetz D, Borysenko L, et al. (2012) Early reactions of Brain-Derived Neurotrophic Factor in plasma (pBDNF) and outcome to acute antidepressant treatment in patients with Major Depression. Neuropharmacology 62: 264-269.
- Xu H, Chen Z, He J, Haimanot S, Li X, et al. (2006) Synergetic effects of quetiapine and venlafaxine in preventing the chronic restraint stress-induced decrease in cell proliferation and BDNF expression in rat hippocampus. Hippocampus 16: 551-559.
- Yoshimura R, Umene-Nakano W, Ueda N, Ikenouchi-Sugita A, Hori H, et al. (2008) Addition of risperidone to sertraline improves sertraline-resistant refractory depression without influencing plasma concentrations of sertraline and desmethylsertraline. Hum Psychopharmacol 23: 707-713.
- Tamburella A, Micale V, Navarria A, Drago F (2009) Antidepressant properties of the 5-HT4 receptor partial agonist, SL65.0155: behavioral and neurochemical studies in rats. Prog Neuropsychopharmacol Biol Psychiatry 33: 1205-1210.
- Nowacka M, Obuchowicz E (2013) BDNF and VEGF in the pathogenesis of stress-induced affective diseases: an insight from experimental studies. Pharmacol Rep 65: 535-546.
- Bergström A, Jayatissa MN, Mørk A, Wiborg O (2008) Stress sensitivity and resilience in the chronic mild stress rat model of depression; an in situ hybridization study. Brain Res 1196: 41-52.
- Sun R, Li N, Li T (2012) VEGF regulates antidepressant effects of lamotrigine. Eur Neuropsychopharmacol 22: 424-430..
- Clark-Raymond A, Halaris A (2013) VEGF and depression: a comprehensive assessment of clinical data. J Psychiatr Res 47: 1080-1087.
- Iga J, Ueno S, Yamauchi K, Numata S, Tayoshi-Shibuya S, et al. (2007) Gene expression and association analysis of vascular endothelial growth factor in major depressive disorder. Prog Neuropsychopharmacol Biol Psychiatry 31: 658-663.
- Kahl KG, Bens S, Ziegler K, Rudolf S, Kordon A, et al. (2009) Angiogenic factors in patients with current major depressive disorder comorbid with borderline personality disorder. Psychoneuroendocrinology 34: 353-357.
- Berent D, Macander M, Szemraj J, Orzechowska A, Galecki P (2014) Vascular endothelial growth factor A gene expression level is higher in patients with major depressive disorder and not affected by cigarette smoking, hyperlipidemia or treatment with statins. Acta Neurobiol Exp (Wars) 74: 82-90.
- Lee BH, Kim YK (2012) Increased plasma VEGF levels in major depressive or manic episodes in patients with mood disorders. J Affect Disord 136: 181-184.
- Ventriglia M, Zanardini R, Pedrini L, Placentino A, Nielsen MG, et al. (2009) VEGF serum levels in depressed patients during SSRI antidepressant treatment. Prog Neuropsychopharmacol Biol Psychiatry 33: 146-149.
- Blumberg HP, Wang F, Chepenik LG, Kalmar JH, Edmiston E, et al. (2008) Influence of vascular endothelial growth factor variation on human hippocampus morphology. Biol Psychiatry 64: 901-903.
- Isung J, Mobarrez F, Nordström P, Asberg M, Jokinen J (2012) Low plasma Vascular Endothelial Growth Factor (VEGF) associated with completed suicide. World J Biol Psychiatry 13: 468-473.
- Warner-Schmidt JL, Duman RS (2007) VEGF is an essential mediator of the neurogenic and behavioral actions of antidepressants. Proc Natl Acad Sci USA 104: 4647-4652.
- Greene J, Banasr M, Lee B, Warner-Schmidt J, Duman RS (2009) Vascular endothelial growth factor signaling is required for the behavioral actions of antidepressant treatment: pharmacological and cellular characterization. Neuropsychopharmacology 34: 2459-2468.
- Allaman I, Fiumelli H, Magistretti PJ, Martin JL (2011) Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology (Berl) 216: 75-84.
- Fournier NM, Duman RS (2012) Role of vascular endothelial growth factor in adult hippocampal neurogenesis: implications for the pathophysiology and treatment of depression. Behav Brain Res 227: 440-449.
- Kempermann G, Kuhn HG, Gage FH (1997) More hippocampal neurons in adult mice living in an enriched environment. Nature 386: 493-495.
- Tanti A, Belzung C (2013) Neurogenesis along the septo-temporal axis of the hippocampus: are depression and the action of antidepressants region-specific? Neuroscience 252: 234-252.
- Mahar I, Bambico FR, Mechawar N, Nobrega JN (2014) Stress, serotonin, and hippocampal neurogenesis in relation to depression and antidepressant effects. Neurosci Biobehav Rev 38: 173-192.
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, et al. (2003) Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 301: 805-809.
- Videbech P, Ravnkilde B (2004) Hippocampal volume and depression: a meta-analysis of MRI studies. Am J Psychiatry 161: 1957-1966.
- Koolschijn PC, van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS (2009) Brain volume abnormalities in major depressive disorder: a meta-analysis of magnetic resonance imaging studies. Hum Brain Mapp 30: 3719-3735.
- Kempton MJ, Salvador Z, Munafò MR, Geddes JR, Simmons A, et al. (2011) Structural neuroimaging studies in major depressive disorder. Meta-analysis and comparison with bipolar disorder. Arch Gen Psychiatry 68: 675-690.
- Chaney A, Carballedo A, Amico F, Fagan A, Skokauskas N, et al. (2014) Effect of childhood maltreatment on brain structure in adult patients with major depressive disorder and healthy participants. J Psychiatry Neurosci 39: 50-59.
- Malberg JE, Eisch AJ, Nestler EJ, Duman RS (2000) Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J Neurosci 20: 9104-9110.
- Mnie-Filali O, Faure C, Mansari ME, Lambás-Señas L, Bérod A, et al. (2007) R-citalopram prevents the neuronal adaptive changes induced by escitalopram. Neuroreport 18: 1553-1556.
- Guilloux JP, Mendez-David I, Pehrson A, Guiard BP, Repérant C, et al. (2013) Antidepressant and anxiolytic potential of the multimodal antidepressant vortioxetine (Lu AA21004) assessed by behavioural and neurogenesis outcomes in mice. Neuropharmacology 73: 147-159.
- Wang Y, Chang T, Chen YC, Zhang RG, Wang HN, et al. (2013) Quetiapine add-on therapy improves the depressive behaviors and hippocampal neurogenesis in fluoxetine treatment resistant depressive rats. Behav Brain Res 253: 206-211.
- Betry C, Etievant A, Pehrson A, Sánchez C, Haddjeri N (2012) Effect of the multimodal antidepressant LuAA21004 on rat cognitive processes. 8th FENS forum of Neuroscience, Barcelona, Spain.
- Snyder JS, Kee N, Wojtowicz JM (2001) Effects of adult neurogenesis on synaptic plasticity in the rat dentate gyrus. J Neurophysiol 85: 2423-2431.
- Ge S, Goh EL, Sailor KA, Kitabatake Y, Ming GL, et al. (2006) GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439: 589-593.
Citation: Bahri S, Mnie-Filali O, Dkhissi-Benyahya O, Abrial E, Haddjeri N (2014) Neuroadaptations of the 5-HT System Induced by Antidepressant Treatments: Old and New Strategies. J Addict Addictv Disord 1: 001.
Copyright: © 2014 Senda Bahri, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.