Perioperative and Postoperative Management in Patients with Alpha-2-Antiplasmin Deficiency: A Case Study of 67-Year-Old Male Undergoing Operative Distal Femur Repair
*Corresponding Author(s):
Gregory MonohanDivision Of Hematology Blood And Marrow Transplant, University Of Kentucky College Of Medicine, United States
Tel:+1 8593235877,
Email:gpmono0@email.uky.edu, monohan@uky.edu
Abstract
Introduction
Alpha-2-antiplasmin deficiency is a rare autosomal recessive condition characterized by increased fibrinolysis which can subsequently lead to uncontrolled hemorrhage. First described in Japan in 1969 by Masateru Kohakura, alpha-2-AP deficiency is rare with fewer than 50 published case studies to date. Individuals with alpha-2-AP deficiency will have normal coagulation parameters including platelet count, bleeding time, Prothrombin Time (PT), activated Partial Thromboplastin Time (aPTT), and clotting factor titers. In alpha-2-AP deficiency patients, clots are able to form normally but degrade in an accelerated fashion prior to the initiation of tissue and vascular repair. Diagnosis of alpha-2-AP deficiency thus requires a high index of suspicion and can be confirmed with specific functional and immunological antiplasmin assays. Here we present a case of a 67-year-old male with known history of homozygous alpha-2-AP deficiency undergoing intramedullary fixation of a distal femur fracture, followed by a discussion of the literature on Perioperative and postoperative strategies for individuals with α2-AP deficiency.
Case Discussion
A 67-year-old male with known history of homozygous alpha-2-AP deficiency presented to our institution with a displaced distal left femur fracture status post a fall from standing height. He has a past medical history of coal workers pneumoconiosis, hypertension, treated hepatitis C infection, and hypothyroidism as well as prior right humeral fracture requiring surgical correction and previous intracranial hemorrhage in 2013 from a horse accident, both successfully managed with antifibrinolyitic therapy. Immediate pre-operative management for bleeding risk included infusion of FFP and 1 gram of tranexamic acid IV bolus. He then received a continuous infusion of tranexamic acid intraoperatively. He underwent uneventful intramedullary fixation of the left distal femur. After completion of the infusion, he was placed on tranexamic acid 1300 mg q 8 hours. Unfortunately, on post-operative day 7, patient developed a new Left Lower extremity deep venous thrombosis despite prophylactic LMWH.
Discussion
Patients with alpha-2-AP much like those with hemophilia have increased bleeding tendencies during surgical procedures. Generally, adequate hemostasis can be managed with administration of anti-fibrinolytic agents. As in our case, there remains the potential for thrombotic events related to antifibrinolyitic therapy and the benefits versus risks of treatment will need to be considered in every surgical case in patients with diagnosis of homozygous alpha-2-AP deficiency. Since it is a rare entity, there is limited medical literature to utilize as guidelines in managing such patients and the approach to post-operative hemorrhaging and DVT prophylaxis in individuals with defects in the fibrinolysis pathway should be considered on a case-by-case basis taking into consideration additional risk factors for both bleeding and thrombosis.
Keywords
INTRODUCTION
The formation of a fibrin clot represents the final step in secondary hemostasis representing a common end point of the intrinsic and extrinsic coagulation pathways [18]. The fibrin meshwork exists in a delicate equilibrium controlled by factors promoting fibrin formation as well as the reverse process of fibrinolysis, which controls clot formation to a minimum necessary to facilitate tissue repair. Tissue type Plasminogen Activator (tPA) and urokinase type Plasminogen Activator (uPA) play primary roles in fibrinolysis by converting protein plasminogen to an active serine protease, plasmin. Plasmin function is to cleave fibrin and is inhibited by α2-AP [19]. This interplay is depicted in figure 1. α2-AP forms a direct complex with plasmin via factor XIIIa, inhibits adsorption of plasminogen to fibrin, and strengthens the local fibrin network through cross linking. α2-AP is mainly synthesized in the liver, but some production ha been observed in the kidney and brain. It has a half-life of 2.6 days [1]. Acquired α2-AP can be seen in liver disease, amyloidosis, and in iatrogenic states during fibrinolytic therapy [16]. Bleeding episodes in those with phenol typically significant α2-AP are typically well controlled with oral antifibrinolyitic agents, such as aminocaproic acid or tranexamic acid which act by preventing plasminogen from binding fibrin [1].
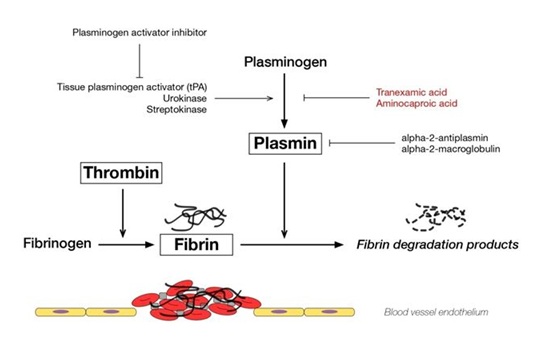
Figure 1: Promoters and inhibitors of fibrinolysis. Alpha-2-antiplasmin regulates plasmin to inhibit fibrinolysis.
Patients with α2-AP deficiency represent unique challenges in the operating room and during recovery from surgical procedures. Here we present a case of a 67-year-old male with known history of homozygous α2-AP deficiency undergoing intramedullary fixation of a distal femur fracture, followed by a discussion of the literature on perioperative and postoperative strategies for individuals with α2-AP deficiency.
CASE DISCUSSION
Patient weighed 111 kilograms at presentation. As expected, our patient had normal coagulation laboratory tests at admission. His I.N.R. was 1.1 with a Prothrombin Time (PT) of 14.1 seconds and an activated Partial Thromboplastin Time (aPTT) of 30 seconds. Immediate pre-operative management for bleeding risk included infusion of two units of FFP and 1 gram of tranexamic acid IV bolus over 10 minutes (100 mg/mL infused at 1 mL/minute). Post infusion coagulation parameters remained normal. It would have been ideal to have α2-AP activity levels before, during, and after the treatment; however, the testing for α2-AP activity is not a readily available test and could not be medically justified. He then received a continuous infusion of1 gram of tranexamic acid intraoperatively (100 mg/mL infused at 1.25 mL/hour for 8 hours). He underwent uneventful intramedullary fixation of the left distal femur. After completion of the infusion, he was placed on tranexamic acid 1300 mg q 8 hours. Patient was also placed on Low-Molecular Weight Heparin (LMWH) 30 mg twice a day for Deep Venous Thrombosis (DVT) prophylaxis. He demonstrated good hemostasis with no postoperative hemorrhaging and was discharged in stable condition on post-operative day 2 to a rehabilitation facility.
Unfortunately, on post-operative day 7, patient developed a new Left Lower extremity deep venous thrombosis despite prophylactic LMWH. At that time, his LMWH dose was increased to 100 mg bid with continuation of home dose tranexamic acid until repeat duplex ultrasound obtained post-operative day 11 demonstrated no evidence of thrombotic extension. His tranexamic acid was discontinued at that time and he was treated with prophylactic LMWH 30 mg BID for a total of six weeks. Follow up duplex ultrasound after therapy showed no evidence of residual thrombosis.
DISCUSSION
Shahian and Levine reported a case of open aortic valve replacement and coronary bypass in an individual with known heterozygous α2-AP deficiency with serum α2-AP concentration of 52 percent. Patient underwent infusion of 3000 mL of FFP, which increased perioperative α2-AP levels to 78 percent [6]. Morimoto et al describes management of intraoral bleeding in those with α2-AP deficiency with 7.5-10 mg/kg of tranexamic acid orally every 6 hours starting 3 hours before procedure to be continued for 7 days [20]. There are no published recommendations for aminocaproic acid dosing in this setting. As recommended by our Hemophilia Treatment Center, our patient received two units of FFP and 1g of IV tranexamic acid bolus pre-operatively with a continuous infusion of 1g tranexamic acid over eight hours intraoperatively and transition to oral tranexamic acid 1300 mg q 8 hours. The incidence of thrombosis and pulmonary embolism is not known for aminocaproic acid administration, but for tranexamic acid, the incidence for either entity is < 1% [21].
The guidelines for DVT prophylaxisin patients with α2-AP deficiency are also not established given the rarity of this condition. In xenograft models of Pulmonary Emboli (PE), α2-AP was shown to play a critical role in resistance to thrombolysis [22] and its inhibitionled to thrombolysis in a comparable manner to tPA administration [23], perhaps even with synergistic effects with tPA in decreasing mortality from PE [24]. These findings open the possibility of utilizing α2-AP in a therapeutic capacity for thrombotic events. Despite the antithrombotic tendencies inherent in those with α2-AP deficiency, orthopedic procedures on weight bearing bones and joints in particular result in higher postoperative risk of DVT given the likelihood of post-operative immobility. Similarly, in patients with hemophilia, pharmacologic thromboprophylaxis is also considered controversial. At least one source does not recommended thromboprophylaxis for those hemophilia patients with aninhibitor given the high risk of bleeding but does recommended thromboprophylaxis after orthopedic procedures in those without inhibitor [25].
In our patient, prophylactic LMWH at 30g bid in conjunction with tranexamic acid was sufficient in preventing post-operative hemorrhage, but was unsuccessful in preventing DVT. It is unknown whether full dose therapeutic LMWH would have been more successful in preventing the DVT without compromising post-operative bleeding risk. Heparin products work by binding antithrombin and accelerating its activity by 1000 fold. The result is decrease in Factor Xa and IIa (activated Thrombin) influence in the coagulation system. This seemingly predates the formation of Fibrin and hence the fibrinolytic pathway as well. In reality, these opposing forces that balance hemostasis are occurring simultaneously. In α2-AP deficiency, Plasmin is left activated and able to degrade Fibrin leading to clot dissolution. Inhibition of the fibrinolytic pathway with tranexamic acid can lead to unopposed fibrin longevity with possible propagation to pathologic thrombosis. The approach to post-operative DVT prophylaxisin individuals with defects in the fibrinolysis pathway should be considered on a case-by-case basis taking into consideration additional risk factors for bleeding and thrombosis such as thrombocytopenia, extend of surgical trauma, immobility, malignancy, obesity, and concurrent medications. Recognizing that both entities are increased in this patient population is paramount. Although this situation is rare, it is our practice to utilize anti-fibrinolytic agents to correct the known hereditary defect while administering standards of care anticoagulation to minimize the thrombotic risk. As this case demonstrates, even with careful consideration, morbidity can occur.
REFERENCES
- Carpenter SL, Mathew P (2008) Alpha2-antiplasmin and its deficiency: fibrinolysis out of balance. Haemophilia 14: 1250-1254.
- Aoki N, Saito H, Kamiya T, Koie K, Sakata Y, et al. (1979) Congenital deficiency of alpha 2-plasmin inhibitor associated with severe hemorrhagic tendency. J Clin Invest 63: 877-884.
- Dawley B (2011) Alpha II Antiplasmin Deficiency Complicating Pregnancy: A Case Report. Obstet Gynecol Int 698648.
- Harish VC, Zhang L, Huff JD, Lawson H, Owen J (2006) Isolated antiplasmin deficiency presenting as a spontaneous bleeding disorder in a 63-year-old man. Blood Coagul Fibrinolysis 17: 673-675.
- Miyauchi Y, Mii Y, Aoki M, Tamai S, Takahashi Y (1996) Operative Treatment of Intramedullary Hematoma Associated with Congenital Deficiency of alpha sub 2-Plasmin Inhibitor, A Report of Three Cases. J Bone Joint Surg Am 78: 1409-1414.
- Shahian DM, Levine LD (1990) Open-Heart Surgery in a Patient with Heterozygous α2-Antiplasmin Deficiency: Perioperative Strategies in the First Reported Case. Chest 97: 1488-1490.
- Griffin GC, Mammen EF, Sokol RJ, Perrotta AL, Stoyanovich A (1993) Alpha 2-antiplasmin deficiency. An overlooked cause of hemorrhage. Am J Pediatr Hematol Oncol 15: 328-330.
- Paqueron X, Favier R, Richard P, Maillet J, Murat I (1997) Severe postadenoidectomy bleeding revealing congenital alpha2 antiplasmin deficiency in a child. Anesth Analg. 84: 1147-1149.
- Hayward CP, Cinà CS, Staunton M, Jurriaans E (2005) Bleeding and thrombotic problems in a patient with alpha2 plasmin inhibitor deficiency. J Thromb Haemost 3: 399-401.
- Miles LA, Plow EF, Donnelly KJ, Hougie C, Griffin JH (1982) A bleeding disorder due to deficiency of alpha 2-antiplasmin. Blood 59: 1246-1251.
- Devaussuzenet VM, Ducou-le-Pointe HA, Doco AM, Mary PM, Montagne JR, et al. (1998) A case of intramedullary haematoma associated with congenital α 2-plasmin inhibitor deficiency. Pediatr Radiol 28: 978-980.
- Kluft C, Vellenga E, Brommer EJ, Wijngaards G (1982) A familial hemorrhagic diathesis in a Dutch family: an inherited deficiency of alpha 2-antiplasmin. Blood 59: 1169-1180.
- Kettle P, Mayne EE (1985) A bleeding disorder due to deficiency of alpha 2-antiplasmin. Journal of clinical pathology 38: 428-429.
- Yoshinaga H, Hirosawa S, Chung DH, Miyasaka N, Aoki N, et al. (2000) A novel point mutation of the splicing donor site in the intron 2 of the plasmin inhibitor gene. Thrombosis and haemostasis 84: 307-311.
- Kordich L, Feldman L, Porterie P, Lago O (1985) Severe hemorrhagic tendency in heterozygous alpha 2-antiplasmin deficiency. Thromb Res 40: 645-651.
- Abdul S, Leebeek FW, Rijken DC, Uitte de Willige S (2016) Natural heterogeneity of α2-antiplasmin: functional and clinical consequences. Blood 127: 538-545.
- Favier R, Aoki N, de Moerloose P (2001) Congenital α2?plasmin inhibitor deficiencies: a review. Br J Haematol 114: 4-10.
- Leavitt AD, Minichiello T (2018) Disorders of Hemostasis, Thrombosis, & amp; Antithrombotic Therapy. Current Medical Diagnosis & Treatment 2018, McGraw-Hill Education, New York, USA.
- Aoki N (2005) Discovery of α2?plasmin inhibitor and its congenital deficiency. Journal of Thrombosis and Haemostasis 3: 623-631.
- Morimoto Y, Yoshioka A, Imai Y, Takahashi Y, Minowa H, et al. (2004) Haemostatic management of intraoral bleeding in patients with congenital deficiency of α2?plasmin inhibitor or plasminogen activator inhibitor?1. Haemophilia 10: 669-674.
- Ferring Pharmaceuticals (2019) Lysteda: Package Insert and Label Information. Ferring Pharmaceuticals Inc, New Jersey, USA.
- Butte AN, Houng AK, Jang IK, Reed GL (1997) Antiplasmin Causes Thrombi to Resist Fibrinolysis Induced by Tissue Plasminogen Activator in Experimental Pulmonary Embolism. Circulation. 95: 1886-1891.
- Singh S, Houng A, Reed GL (2017) Releasing the Brakes on the Fibrinolytic System in Pulmonary Emboli: Unique Effects of Plasminogen Activation and α2-Antiplasmin Inactivation. Circulation 135: 1011-1020.
- Matsuno H, Okada K, Ueshima S, Matsuo O, Kozawa O (2003) Alphaα2?Antiplasmin plays a significant role in acute pulmonary embolism. Journal of thrombosis and haemostasis 1: 1734-1739.
- Ozelo MC (2012) Surgery in patients with hemophilia: is thromboprophylaxis mandatory? Thromb Res 130 : 23-26.
Citation: Nakazawa M, Monohan G (2019) Perioperative and Postoperative Management in Patients with Alpha-2-Antiplasmin Deficiency: A Case Study of 67-Year-Old Male Undergoing Operative Distal Femur Repair. J Hematol Blood Transfus Disord 6: 023.
Copyright: © 2019 Mary Nakazawa, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.