Peripheral Biomarkers of Tobacco-Use Disorder: A Systematic Review
*Corresponding Author(s):
Dwight F NewtonDepartment Of Pharmacology And Toxicology, University Of Toronto, Toronto, Ontario, Canada
Tel:+1 6479864337,
Email:dwight.newton@mail.utoronto.ca
Abstract
Tobacco Use Disorder (TUD) is a major worldwide healthcare burden resulting in 7 million deaths annually. TUD has few approved cessation aids, all of which are associated a high rate of relapse within one year. Biomarkers of TUD severity, treatment response, and risk of relapse have high potential clinical utility to identify ideal responders and guide additional treatment resources.
Methods
A MEDLINE search was performed using the terms biomarkers, dihydroxyacetone phosphate, bilirubin, inositol, cotinine, adrenocorticotropic hormone, cortisol, pituitary-adrenal system, homovanillic acid, dopamine, pro-opiomelanocortin, lipids, lipid metabolismall cross-referenced with tobacco-use disorder.
Results
The search yielded 424 results, of which 57 met inclusion criteria. The most commonly studied biomarkers were those related to nicotine metabolism, the Hypothalamic-Pituitary-Adrenal (HPA) axis, and Cardiovascular (CVD) risk. Nicotine metabolism was most associated with severity of dependence and treatment response, where as HPA axis and CVD markers showed less robust associations with dependence and relapse risk.
Conclusion
Nicotine-metabolite ratio, cortisol, and atherogenicity markers appear to be the most promising lead biomarkers for further investigation, though the body of literature is still preliminary. Longitudinal, repeated-measures studies are required to determine the directionality of the observed associations and determine true predictive power of these biomarkers. Future studies should also endeavour to study populations with comorbid psychiatric disorders to determine differences in utility of certain biomarkers.
Keywords
INTRODUCTION
Biological markers of TUD hold great promise to address these problems which are pervasive across psychiatric disorders. Current diagnostic measures are based on identifying behavioural symptoms (e.g. loss of control, craving) and essentially no objective biologically-informed treatment selection tools exist [6]. However, it is well understood that the underlying biology of TUD involves neuroplastic changes in reward circuitry occurring with repeated drug use and the formation of drug-salient environmental and interoceptive cues [7]. Maladaptations of the Dopamine (DA) system of the nucleus accumbens and ventral tegmental area occurring in addictive disorders have been extensively characterized, and drugs of abuse almost invariably induce changes in this circuitry with repeated use [8].
Neuroimaging has provided a window to study these structural and functional changes in humans, and identify biomarkers related to specific symptom dimensions, such as craving or impulse control, and clinical phenomena such as relapse and treatment response [8-10]. Despite their immediate relevance to the pathophysiology of TUD, Central Nervous System (CNS)-based biomarkers are particularly costly to acquire and require specialized facilities and technical staff, greatly limiting their scalability and utility as predictive biomarkers. Such considerations are especially relevant when biomarkers are measured repeatedly over time, in the case of markers of treatment efficacy or relapse or in healthcare settings without neuroimaging equipment. Peripheral (non-CNS) markers, though potentially lacking direct representations of CNS pathophysiology, have the advantages of being easily collected, inexpensive, and can be rapidly integrated into ongoing clinical practice.
Recent reviews of biomarkers in addictive disorders have largely focused neuroimaging markers, which is warranted given the known similarity of neurobiological changes occurring in the mesolimbic DA system across drugs of abuse [8,11,12]. Reviews of tobacco-using populations specifically have been limited by either focusing on all smoking populations without consistently quantified levels of dependence or examining a single class of biomarkers (e.g. nicotine metabolites) [13-16]. Genetic markers, while clearly relevant to TUD clinical outcomes [17-19], have limited a ability to track dynamic changes over time. Dynamic, non-genetic, biomarkers are better suited to capturing features which change over time such as the course of treatment response or risk of relapse. A focus on TUD may be more clinically relevant than examining smokers regardless of dependence and tobacco use intensity. Though nicotine induces changes in nicotinic receptor expression after a single dose which persists after clearance of the drug, the neurobiological changes occurring in addiction involve systems beyond the specific pharmacology of the drug of abuse [20]. Furthermore, a focus on TUD may capture a more severe subgroup of the tobacco-using population which may decrease the potential noise in biomarker signals occurring in less dependent groups.
Existing biomarkers for TUD, and SUDs more broadly, have generally been those related to drug exposure [6]. In the case of nicotine, these have been expired Carbon Monoxide (CO) and cotinine measured in saliva, urine, or blood [6]. Expired CO is increased acutely (6-9 hours) after combustible tobacco use, whereas cotinine, the primary metabolite of nicotine, has a longer half life (5-7 days) thus giving a longer-term measure of exposure to nicotine, regardless of the route of administration [21]. However, growing evidence suggests that these, other exposure markers, are associated with dependence severity and risk of relapse [22,23]. Biomarkers not directly related to nicotine metabolism have been increasingly studied in TUD, such as markers of Cardiovascular Disease (CVD), inflammation, oxidative stress, and Hypothalamic-Pituitary-Adrenal (HPA) axis reactivity [24-26]. Such markers may be related to the underlying pathophysiology of TUD or relevant comorbidities, and thus may relate more strongly or specifically to symptom dimensions or relevant clinical outcomes.
Given the growth of the literature regarding biomarkers related to smoking and tobacco use, and the broader, or distinct, scope of existing reviews, a systematic review of the extant literature of peripheral biomarkers related to TUD will be performed. This review will focus on TUD and nicotine dependence specifically and aims to determine the peripheral biomarkers related to TUD and relevant clinical outcomes. Suggestions regarding future research directions and clinical implications will also be presented.
RESULTS
Search results and study selection
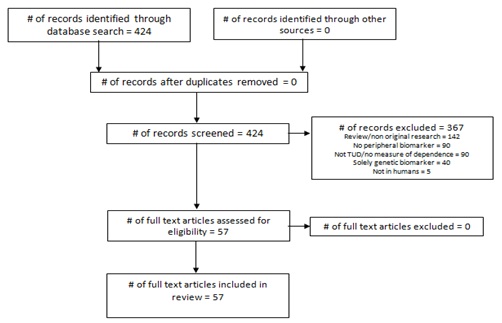
|
Study |
Sample |
Biomarker Class |
Outcome Measures |
|
Jacob et al, 2004 [27] |
3 cohorts (1) Smokeless tobacco users: 50 NRT, 50 placebo (intake, 6 weeks); (2) Smokeless tobacco users: 55 NRT+therapy, 50 placebo+therapy, 51 NRT only, 54 placebo only; (3) 99 smokers |
Exposure |
Urinary alkaloids (anabasine, anatabine, and nornicotine), nicotine, and cotinine |
|
Carim-Todd et al, 2016 [28] |
Smokers: 63 education +NRT+ support, 63 education + support, 64 education only. (intake, 1, 2, 3, 4, 12 wks) |
Exposure |
Urinary cotinine |
|
Frost-Pineda et al, 2014 [14] |
1768 smokers |
Exposure |
Expired CO |
|
McPherson et al, 2014 [29] |
103 non-treatment seeking smokers |
Exposure |
Salivary cotinine |
|
Jung et al, 2012 [30] |
381 smokers |
Exposure |
Urinary cotinine |
|
Krishnan-Sarin et al, 1999 [31] |
56 smokers |
Exposure |
Plasma cotinine |
|
Mushtaq et al, 2012 [32] |
95 smokeless tobacco users |
Exposure |
Salivary cotinine |
|
Jao et al, 2017 [22] |
474 smokers undergoing treatment |
Exposure |
NMR |
|
Pacek et al, 2016 [33] |
839 non-treatment seeking smokers switched to VLNCs |
Exposure |
Expired CO, NMR, urinary NNAL |
|
Brandon et al, 2007 [4] |
89 smokers with SCZ, 53 healthy smokers |
Exposure |
Serum nicotine, cotinine, 3-HC |
|
Kaufmann et al, 2015 [34] |
21 male smokers |
Exposure |
Plasma nicotine, urinary cotinine and 3-HC |
|
Fu et al, 2012 [35] |
196 non-treatment seeking smokers |
Exposure |
Salivary cotinine |
|
Kwok et al, 2014 [36] |
1050 pregnant smokers |
Exposure |
Blood cotinine, salivary cotinine, expired CO |
|
Van et al, 2016 [23] |
239 daily smokers |
Exposure |
Urinary nicotine, cotinine, and 3-HC |
|
Benowitz et al, 2009 [37] |
20 non treatment-seeking smokers |
Exposure |
Plasma nicotine, cotinine, carboxy hemoglobin NNAL and other toxicants |
|
St et al, 2013 [38] |
69 black and 135 white smokers |
Exposure |
Urinary TNEs, NNAL, and PAH metabolites |
|
Hatsukami et al, 2010 [39] |
391 smokers |
Exposure |
Urinary nitrosamines, cotinine, and TNE, |
|
Nordskog et al, 2015 [84] |
840 smokers (780 completed) |
Exposure |
Urinary TNE, salivary NMR, urinary nitrosamines |
|
Kaufmann et al, 2015 [34] |
499 treatment-seeking smokers |
Exposure |
salivary NMR |
|
Hatsukami et al, 2010 [39] |
53 - 0.05mg cigarette, 52 - 0.3mg cigarette, 60 4mg nicotine lozenge |
Exposure |
Urinary cotinine, NNAL, NNK, NNN, 1-HOP, PAH, other toxicants |
|
Kawada et al, 2010 [40] |
282 male smokers |
Exposure |
Salivary nicotine and cotinine |
|
Ferketich et al, 2007 [41] |
256 smokeless tobacco users |
Exposure |
Salivary cotinine |
|
Muhammad-Kah et al, 2011 [42] |
3585 smokers |
Exposure |
Urinary TNEs, serum cotinine, carboxy hemoglobin |
|
Komiyama et al, 2013 [43] |
186 smokers |
CVD/Lipids |
Triglycerides, LDL, HDL, CRP |
|
Nunes et al, 2015 [44] |
120 TUD, 77 controls, 134 mood disorders |
CVD/Lipids |
Total cholesterol, triglycerides, HDL, LDL. Atherogenic index of plasma (TG/HDL) and atherogenic coefficient (nonHDL/HDL) |
|
Cao et al, 2014 [45] |
160 moderately dependent smokers (80 treatment, 80 control) |
CVD/Lipids |
Total cholesterol, triglycerides, HDL, LDL |
|
Bortolasci et al, 2015 [46] |
90 MetS (46 TUD) and 224 (82) nonMetS |
CVD/Lipids |
Plasma MDA, HDL, triglycerides |
|
Suhaimi et al, 2016 [47] |
107 Male smokers |
CVD/Lipids |
Serum leptin, lipid profile |
|
Zaparoli et al, 2016 [48] |
63 smokers (RCT) |
CVD/Lipids |
DHA and EPA |
|
Korhonen et al, 2011 [49] |
130 female smokers |
CVD/Lipids |
CRP, WBC/RBC counts, TG, TC, HDL, LDL, TC/HDL ratio |
|
Tanner et al, 2015 [18] |
60 smokers, 48 chewers, 60 controls |
CVD/Lipids |
Urinary TNEs and nitrosamines, Serum nicotine and cotinine |
|
Nordskog et al, 2015 [50] |
60 smokers, 48 chewers, 60 controls |
CVD/Lipids |
Serum lipid profile and hematological markers |
|
Scaglia et al, 2016 [51] |
50 smokers and 50 controls |
CVD/Lipids |
PUFAs in RBCs - alphalinolenic acid, EPA, docosapentaenoic acid, and DHA |
|
Selya et al, 2017 [52] |
3903 adult smokers |
CVD/Lipids |
Cholesterol, HDL, LDL, LDL/HDL ratio |
|
Rasmusson et al, 2006 [53] |
33 smokers |
Stress |
Plasma cortisol , DHEA, DHEA-S, and cotinine |
|
al'Absi et al, 2007 [54] |
72 smokers |
Stress |
Salivary cotinine and cortisol, plasma cortisol and ACTH |
|
al'Absi et al, 2008 [55] |
23 smokers, 25 controls |
Stress |
Salivary cortisol, Plasma cortisol and ACTH |
|
Lee et al, 2005 [56] |
25 smokers (13 naltrexone, 12 placebo) |
Stress |
Plasma beta endorphin, dynorphin A, ACTH, serum cortisol and prolactin |
|
Buchmann et al, 2010 [57] |
50 daily smokers, 17 occasional smokers, 64 controls |
Stress |
Plasma cortisol |
|
Pomerleau et al, 2004 [58] |
12 female smokers with MDD, 12 female healthy smokers |
Stress |
Plasma cortisol and ACTH |
|
Wardle et al, 2011 [59] |
49 smokers (16 smoke as normal, 17 - asbtinence with placebo patch, 16, abstinence with nicotine patch) |
Stress |
Salivary cortisol |
|
Mendelson et al, 2008 [60] |
24 male smokers |
Stress |
Plasma cortisol, ACTH, DHEA, nicotine |
|
Puddey et al, 1984 [61] |
33 cessation and 33 non-cessation smokers |
Stress |
Plasma epinephrine and norepinephrine, serum cortisol and prolactin |
|
Carim-Todd et al, 2016 [28] |
25 non-dependent smokers, 23 heavy smokers, and 25 controls |
Stress |
Salivary cortisol |
|
Krishnan-Sarin et al, 1999 [62] |
9 smokers, 11 controls |
Stress |
Plasma cortisol |
|
von et al, 2010 [63] |
60 smokers in acute (3hr) withdrawal, 64 controls |
Stress |
Plasma orexin, leptin, cortisol, ACTH |
|
Gerra et al, 2016 [24] |
50 ND smokers and 50 controls |
Stress |
Cortisol and ACTH |
|
Hamidovic et al, 2017 [64] |
19 and 37 healthy smokers |
Stress |
Circulating cortisol |
|
Cohen et al, 2004 [65] |
20 male smokers |
Stress |
Salivary cortisol |
|
Morris et al, 2016 [66] |
64 smokers |
Stress |
Salivary cotinine, expired CO, salivary cortisol, salivary alpha-amylase |
|
Hogle et al, 2006 [67] |
20 24hr abstinent smokers, 20 non-abstinent, 20 occcasional smokers, 20 controls |
Stress |
Salivary cortisol |
|
Steptoe et al, 2006 [68] |
112 smokers |
Stress |
Salivary cortisol |
|
al'Absi et al, 2013 [69] |
116 smokers, 38 controls |
Stress |
Salivary cortisol, Plasma cortisol and ACTH |
|
Krause et al, 2018 [70] |
20 TUD, crossover design |
Stress |
Cortisol, ACTH, prolactin |
|
Vargas et al, 2013 [26] |
150 smokers, 191 controls |
Other |
CRP, nitric oxide metaoblites, advanced oxidation end products (AOPP), MDA, fibrinogen, total antioxidant potential |
|
Umene-Nakano et al, 2012 [71] |
20 smokers, 21 controls (untreated) |
Other |
Plasma HVA (DA metabolite) and MHPG (NE metabolite) |
|
Waszkiewicz et al, 2013 [72] |
18 colon cancer patients (10 TUD and AUD), 10 controls |
Other |
Serum beta-galactosidase (higher activtiy in younger tissues, indicative of liver damage and/or tumour growth) |
Markers of exposure
A cross-sectional study of 239 smokers measuring urinary nicotine, cotinine, and 3-HC found that levels of all three of these markers were positively correlated with FTND scores [74]. The Time to First Cigarette (TTFC) item of the FTND was positively correlated with urinary levels of nicotine, cotinine, and 3-HC. However, cotinine and 3-HC were not associated with Cigarettes Per Day (CPD). These results were replicated in a separate study of 196 smokers which measured salivary cotinine only [35]. Cotinine was associated with FTND scores, and the FTND item showing the strongest individual association with cotinine was again the TTFC. Urinary cotinine was also correlated with overall FTND scores in a study of 381 smokers [30]. In this study a Receiver Operating Characteristic (ROC) analysis indicated that urinary cotinine could differentiate highly dependent (FTND≥6) smokers from others with 71% sensitivity and 74% specificity. A large-scale study of 3585 smokers examining urinary nicotine equivalents and serum levels of cotinine and carboxyhemoglobin found that levels of each biomarker were associated with FTND scores, with TTFC showing the strongest correlation of all items [42]. In contrast to the above studies, exposure markers were significantly associated with CPD. In a study of 204 smokers (69 black and 135 white), urinary nicotine equivalents, NNAL and PAH were measured [38]. Surprisingly, nicotine equivalents and NNAL levels were correlated with greater FTND scores in white smokers, but this was not the case for black smokers. This demonstrates the need to continue developing psychometric tools for assessing nicotine dependence, as they may not be equally accurate across populations. A study measuring salivary nicotine and cotinine in 282 male smokers found that cotinine was significantly associated with FTND, but not nicotine, once again indicating the importance of measuring metabolites [40]. These findings demonstrate that nicotine metabolites, especially cotinine, are related to severity of nicotine dependence and specific smoking features, such as the TTFC. Surprisingly, correlations between such recency-of-use markers and measures of intake alone (i.e. CPD) were less consistent.
Longitudinal studies of exposure markers have primarily been secondary analyses of Randomized Controlled Clinical Trials (RCTs) or non-randomized studies of smoking cessation, and thus offer an opportunity to look at potential predictive relationships with treatment response. One such secondary analysis of a 8-weekRCT of Nicotine Replacement Therapy (NRT), in the form of a 21mg/day transdermal nicotine patch, measured the impact of NMR at baseline on treatment response in 474 smokers [23]. Those with a lower NMR (i.e. faster metabolizers) had a decreased likelihood of abstinence at week 8. This finding was replicated in a 8-week RCT of combined NRT (patch) and behavioural counselling which measured salivary NMR in 499 treatment-seeking smokers. It was found that patients with faster NMRs showed lower quit rates at 8-weeks [34]. In the same study, NMR was not associated with any changes in craving or withdrawal symptoms. A third 8-week clinical trial of NRT (patch) measured plasma cotinine at baseline and after 1-week in 56 smokers. Percent cotinine replacement did not correlate with any smoking outcome.
Interestingly, this relationship between NMR and likelihood of cessation is also seen in short-term paradigms. A study using a 5-day contingency-management paradigm, in which smokers were monetarily rewarded for a negative expired CO reading on each of the 5 days, measured salivary cotinine in 103 non-treatment seeking smokers at intake [29]. In this study, cotinine levels were predictive of expired CO levels at all time points, poorer study retention, lower chance of 100% abstinence, and shorter time to first positive CO reading. These studies indicate that nicotine metabolites, NMR especially, may be clinically salient biomarkers and individuals with fast NMRs may require further cessation aids. These findings also suggest an importance for composite biomarkers (e.g. NMR), which seem to relate more robustly to NRT response than cotinine alone. Indeed, NMR is likely particularly relevant to NRT given the fact that nicotine itself is the primary therapeutic agent, more so than non-NRT pharmacotherapy such as varenicline or bupropion.
Though the association between exposure markers and dependence established in cross-sectional studies is compelling, it cannot be determined from such studies if this relationship is modifiable. Studies which experimentally modulate nicotine exposure can, however. A secondary analysis of a 6-week RCT investigating Very Low Nicotine Content Cigarettes (VLNCs) (0.4-2.4mg/g nicotine) measured salivary NMR and urinary NNAL at intake, 2-weeks, and 6-weeks [33]. Subjects randomized to VLNCs had lower NNAL and FTND scores after 6-weeks. These findings were broadly replicated in another study of VLNCs, in which 20 non-treatment seeking smokers were progressively tapered to a 0.1mg nicotine VLNC over 6-weeks [37]. Subjects showed reduced FTND scores, cotinine, cHb, and NNAL at 6-weeks compared to baseline, despite the presence of compensatory smoking. cHb and NNAL, but not cotinine, remained decreased at 10-weeks. A similar approach was used in a6-week semi-blinded RCT of VLNCS in which subjects were randomized to either 0.05mg nicotine VLNC (n=53), 0.3mg VLNC (n=52), or a 4mg nicotine lozenge (n=60) in which urinary cotinine, NNAL, NNK, NNN, and PAH were measured at 2 and 6-weeks [39]. Patients receiving 0.05mg VLNCs or lozenges showed reduced nitrosamines levels at 6-weeks compared to the 0.3mg group, and the 0.05mg VLNC group had lower cotinine at 2 and 6-weeks compared to both groups. Though this last study not directly measure dependence, these findings show that reducing tobacco (and nicotine) intake via VLNCs can reduce markers of nicotine metabolites, carcinogenic tobacco-derived nitrosamines, and severity of nicotine dependence. Though it is unsurprising that nitrosamines and other nicotine metabolites are decreased after VLNC substitution, it is notable that their decrease is correlated to, or coincides with, decreased dependence. Lastly, though the above studies were performed in otherwise smoking populations, and these markers maintain correlations with dependence in smokeless tobacco users [32,41], and pregnant smokers [36].
Stress response
Most studies measuring biomarkers of stress have used psychosocial or pharmacological paradigms to induce stress responses, and then determine the relationships between stress hormone levels and clinical outcomes. The Trier Social Stress Test (TSST) has been widely used to induce stress, which involves a public speaking task and surprise mental arithmetic task performed for an unsupportive audience [80]. A study of 64 smokers measured salivary cortisol and alpha-amylase before, and during the response to, the TSST [66]. Those with low nicotine dependence (median split of FTND scores) showed similar baseline cortisol levels to those with high dependence, but showed a greater increase in cortisol in response to the TSST. Highly dependent smokers showed a blunted cortisol response, i.e. no increase after the TSST. This blunted cortisol response has been further replicated in 23 dependent smokers [28], 23 non-dependent smokers, and 25 controls (salivary cortisol), 116 smokers and 38 controls (plasma cortisol) [69], 50 daily smokers and 17 non-daily, and 64 controls (plasma cortisol) where cortisol levels also correlated with craving [57]. Two stress-response studies reported negative findings, which employed fear-conditioning [67] and mood induction [58], paradigms to induce stress.
Similar results have been seen using pharmacological stressors such as opioid antagonists. A study employing a pharmacological challenge, using the opioid antagonist naltrexone, in 23 smokers and 25 controls found that both plasma cortisol and ACTH responses were blunted in smokers compared to controls [55]. Unsurprisingly, peak ACTH response occurred before peak cortisol response, though salivary cortisol levels were not blunted in the smoking group. A blunted plasma cortisol response to naloxone, another opioid antagonist, was also seen in a study of 9 smokers and 11 controls [62]. However, a third naltrexone study did not replicate such findings. Negative results were reported in a study of a dexamethasone challenge to 24 female smokers [56]. Overall, these studies indicate that nicotine dependent smokers show blunted physiological responses to stress, across different stress-induction methods, and a potential implication of the opioid system in some of these changes.
Studies of stress response in smokers have also examined effects of acute abstinence on stress responses, where cortisol responses appear to be less robust. A study of 16 non-abstinent, 17 overnight-abstinent (with nicotine patch), and 16 overnight-abstinent (placebo patch) smokers found blunted TSST-induced salivary cortisol responses in the non-abstinent and nicotine patch groups compared to placebo [59]. No relationships with subjective effects or symptoms were found. A study of 72 smokers undergoing 24-hour abstinence measured salivary cortisol, and plasma ACTH and cortisol responses to a different stress test, the Paced Auditory Serial Addition Task (PASAT) [54]. The stress protocol increased both ACTH and cortisol, though only ACTH showed a blunted response. A recent study of 20 smokers used a crossover design to determine changes in experimentally-induced craving and blood levels of cortisol, ACTH, and prolactin in response to a 1.6mg or 3.2mg naltrexone challenge [70]. Experimental procedures consisted of 4-hour induced abstinence followed by challenge with naltrexone or placebo. Drug administration was followed by smoking-related visual cues or neutral cues. Smoking cues resulted in increased self-reported craving scores. Both naloxone doses resulted in greater levels of cortisol, ACTH, and prolactin in response to smoking-cues, compared to placebo treatment and smoking cues, suggesting a potentiation of the stress response due to cue reactivity during withdrawal. Interestingly, the relationship between cortisol changes and craving may be dissociable, as studied in 37 overnight-abstinent smokers. This study examined the effect of intranasal insulin pre-treatment on blood cortisol levels in response to the TSST [64]. Intranasal insulin reduced measures of craving (visual analog scale) and increased pre-TSST cortisol levels which remained elevated after the TSST. Given the potential utility of cortisol responses clinically, it is important to characterize such cases where associations between biomarkers and clinical outcomes are less robust, and the impact of cessation aids.
The impact of longer-term abstinence was assessed in a group of 33 smokers attempting an 8-day period of abstinence, with follow-up assessment 7-days afterwards to assess relapse [53]. Plasma cortisol, dehydroepiandrosterone (DHEA-an adrenal-derived steroid hormone), DHEA-sulphate, and cotinine were measured. No individual marker was altered by abstinence at 8-days, though the DHEA: Cortisol ratio was lower in those who relapsed at 15 days compared to those who continued abstinence. The DHEA: Cortisol ratio has been proposed to more accurately assess net glucocorticoid production given the opposing actions of DHEA and cortisol [81].
Stress hormones
Two studies have examined longer-term withdrawal in the form of cessation trials. A no-treatment cessation trial measured serum cortisol, catecholamines, and prolactin in 33 quit-attempters and 33 non-quit-attempting smokers at intake and after 6-weeks [61]. Successful quitters showed decreased cortisol and epinephrine, but no baseline measures alone were predictive of a successful quit attempt. The second study, a 6-week NRT (patch) RCT of 112 smokers, measured salivary cortisol at baseline, 1 day, and 1, 2, and 6-weeks [68]. Subjects showed decreased cortisol after 1 day, with successful quitters showing a persistent decrease. Contrary to expectations, greater cortisol decrease after 1 day was associated with greater chance of relapse. These studies show compelling results for the use of cortisol as a predictive marker of treatment response and risk of relapse.
CVD and lipid biomarkers
Commonly reported CVD risk biomarkers include the lipid profile: Total Cholesterol (TC), high-Density Lipoprotein Cholesterol (HDL), Low-Density Lipoprotein Cholesterol (LDL), and Triglycerides (TG) of which TC, LDL, and TG are associated with increased CVD risk and HDL is associated with decreased risk. Additional markers measured in the following studies include Polyunsaturated Fats (PUFAs) which are generally thought to be protective [85] and two calculated risk factors; atherogenic index of plasma (TG:HDL) and the atherogenic coefficient (non-HDL:HDL) [86,87].
Results from the National Health and Nutrition Survey (NHANES) of 3903 current smokers measuring lipid profile found that an earlier TTFC was associated with lower HDL, greater LDL, and a poorer overall lipid profile (i.e. greater percentage of markers in unhealthy ranges) [87]. Other research has found similar results with atherogenic measures, namely a study examining TG, HDL and LDL in 120 patients with TUD alone, 134 patients with comorbid TUD and Mood Disorders (MDD or BD), and 77 controls [44]. Atherogenic index and atherogenic coefficient were both increased in TUD versus controls and were highest in comorbid TUD and mood disorders. Another study, focusing on comorbid disorders, measured plasma Malondialdehyde (MDA - a marker of lipid peroxidation and oxidative stress), HDL, and TG in 90 metabolic syndrome patients (48 with TUD) and 224 non-metabolic syndrome controls (82 with TUD) [46]. Subjects with TUD showed a greater atherogenic index compared to non-TUD subjects, regardless of metabolic syndrome status. Lastly, a cross-sectional study of 107 male smokers found that serum leptin levels, a hormone which stimulates lipolysis, were negatively correlated with FTND scores, though lipid profile measures did not correlate with any TUD measure [47].
Longitudinal studies have expanded on these cross-sectional findings, indicating potential predictive value of CVD markers. A 90-day RCT of a 3mg daily omega-3 PUFA supplement in 63 smokers measured baseline circulating levels of the omega-3 PUFAs Docosahexaenoic Acid (DHA) and Eicosapentaenoic Acid (EPA) [48]. A pre-treatment comparison with 51 controls found decreased DHA levels in smokers, and PUFA supplementation resulted in decreased FTND scores at 90 days though no correlational analyses were performed. A 12-week longitudinal study of cessation-induced weight gain measured lipid profile and CRP in 186 treatment-seeking smokers, 89 receiving 21mg nicotine patch and 95 receiving varenicline [43]. At baseline and 12-weeks, BMI positively correlated with TG levels and FTND scores, and negatively with HDL. Greater baseline FTND scores and TG levels were predictive of BMI gain at 3 months. BMI changes are notable in this study, as cessation-related weight gain is a well-documented risk factor for medication non-compliance and unsuccessful quit attempts [88]. Lastly, a 12-week RCT of exercise therapy assessed lipid profile, red and white blood cell concentration, and C-reactive protein (CRP - a marker of inflammation) in 130 female smokers at baseline and at 12-weeks [49]. Those who remained abstinent at 12-weeks had reduced TC and TC/HDL ratio, and greater baseline CRP levels were predictive of relapse at 12-weeks.
Negative findings, or no associations with dependence were performed, in 3 studies. A cross-sectional study of 60 smokers, 48 smokeless tobacco users, and 60 non-tobacco users measuring lipid profile and a panel of hematological markers [50]. Another cross-sectional study measuring circulating PUFA levels in 50 smokers and 50 non-smokers [51]. Lastly no associations between FTND scores and lipid profile were found in a 12-month RCT of atorvastatin treatment in 160 smokers [45].
Overall, these results suggest that atherogenic markers show more robust associations with severity of dependence and successful treatment responses (i.e. maintenance of abstinence or fewer adverse effects) than individual markers. Though these markers are disrupted in TUD, perhaps suggestive of utility as diagnostic aids, specific associations with measures of dependence or other symptom dimensions are understudied. Thus, the evidence is more tentative for these markers, though they represent an interesting research opportunity given their potential as dual-use biomarkers, for both CVD and TUD outcomes.
Other markers
DISCUSSION
Stress marker results suggest they have utility being measured at steady-state levels over time, and in response to a stress challenge. More severely dependent smokers showed more blunted cortisol and ACTH responses [28, 54,55, 57,68,69], which may be specific to the presence of nicotine [59,67], and may indicate a disrupted ability to cope with stressful situations. Cortisol responses to stress may have potential use as a diagnostic aid and longitudinal index of dependence, though this requires a validated and repeatable stress-induction procedure, as no study above performed the TSST more than once. Less consistent results were found with naloxone/naltrexone challenges, with positive and negative findings reported [31,56]. Furthermore, the importance of a standardized stress procedure was highlighted by more robust associations being found in studies using the TSST, versus more negative findings in studies using other psychosocial stressors. Lastly, only one study examined stress hormones as predictive markers of treatment response, though positive findings were observed regarding their prediction of relapse [53]. Future studies should examine the utility of these markers prospectively to properly characterize whether baseline stress hormone levels, or stress-induced levels, are predictive of treatment response.
CVD markers showed promising results, though the body of literature was considerably smaller compared to stress or exposure markers. Severity of dependence was associated with less favourable lipid profiles [88], with HDL, LDL, and atherogenicity measures being most robustly associated with a TUD diagnosis [46,89]. TG and PUFA levels showed tentative associations with treatment response, however these were reported in single studies and require replication [43,45,48]. Tentative evidence was also observed for the diversity of CVD markers measured in other studies (i.e. leptin, CRP, hemodynamic measures) [48,49,50]. Those markers classified as “others” were underpowered and in highly confounded populations (colon cancer) when positive findings were found [72], or did not include relevant predictive analyses [71].
These findings should be interpreted in the context of the limitations of this review. First, by limiting the search to TUD and nicotine dependence, some relevant studies measuring dependence but not categorized under the MeSH term were inevitability excluded. This was done given the impracticability of including broad terms such as smoking (the inclusion of which added nearly 5,000 abstracts), and the desire to not capture an inordinate number of studies not reporting dependence measures. Second, some potential biomarkers may have been missed, as they may not have fallen under the general biomarker term in the search. However, given the commonly reported markers in other biomarker reviews [5,6], this number was likely small. Third, though this systematic review endeavoured to study TUD and dependence in an objective manner, the information provided by the 6-item FTND, the primary dependence scale, limits the aspects of dependence able to be studied. Further development of self-report and interview-based tools for assessing dependence is warranted.
Overall, these findings indicate that cotinine, NMR, and stress markers (cortisol and ACTH) emerge as lead biomarkers of dependence and treatment response, and prospective studies of their clinical utility are warranted. Longitudinal studies of other biomarkers, particularly CVD markers, presented here should be performed, given the preponderance of cross-sectional study designs and their inability to determine directionality of associations. Studies integrating genetics and neuroimaging markers with peripheral biomarkers were not found in the literature search and would improve the understanding of how these peripheral biomarkers relate to and interact with genetic predispositions and the underlying neurobiology of TUD. Importantly, the absence of a mechanistic understanding of how a peripheral biomarker relates to the CNS need not prevent its use clinically, if it shows a robust correlation with clinically relevant measures.
Finally, this review found that composite biomarkers (e.g. NMR, atherogenic indices) showed, generally, more robust results and greater predictive power than single biomarkers. Future studies should further this trend and examine novel composite biomarkers through statistical methods (e.g. propensity scoring, ROC, principal component analyses, or machine learning approaches) and a priori knowledge that best predict symptom severity, treatment response, and risk of relapse. Such studies will improve the treatment of TUD and may allow personalized prognoses and therapies based on biomarker profiles.
METHODS
Studies were included if they reported quantitative measures of peripheral biological compounds in individuals diagnosed with TUD, objectively measured as nicotine dependent, or undergoing cessation treatment. Studies without human subjects, those with SUD populations that did not report TUD findings independently, or those without original research findings (e.g. reviews or meta-analyses) were excluded. Studies whose biological markers were solely genetic were also excluded, given the focus of this review on dynamic biomarkers. Studies measuring only expired CO were not included if this measure was used solely for assessing smoking status.
REFERENCES
- World Health Organization (2018) Tobacco. World Health Organization, Geneva, Switzerland.
- American Psychiatric Association (2013) Diagnostic and statistical manual of mental disorders (DSM-5). American Psychiatric Association, Washington, D.C., USA.
- American Academy of Addiction Psychiatry (2015) Nicotine dependence. American Academy of Addiction Psychiatry, East Providence, USA. Pg no: 1-4.
- Brandon TH, Vidrine JI, Litvin EB (2007) Relapse and relapse prevention. Annu Rev Clin Psychol 3: 257-284.
- Camenga DR, Klein JD (2016) Tobacco Use Disorders. Child Adolesc Psychiatr Clin N Am 25: 445-460.
- Bough KJ, Pollock JD (2018) Defining Substance Use Disorders: The Need for Peripheral Biomarkers. Trends in molecular medicine 24: 109-120.
- Volkow ND, Morales M (2015) The Brain on Drugs: From Reward to Addiction. Cell 162: 712-725.
- Fedota JR, Stein EA (2015) Resting-state functional connectivity and nicotine addiction: Prospects for biomarker development. Ann N Y Acad Sci 1349: 64-82.
- Menossi HS, Goudriaan AE, de Azevedo-Marques Perico C, Nicastri S, de Andrade AG, et al. (2013) Neural bases of pharmacological treatment of nicotine dependence - insights from functional brain imaging: A systematic review. CNS drugs 27: 921-941.
- Sutherland MT, Stein EA (2018) Functional neurocircuits and neuroimaging biomarkers of tobacco use disorder. Trends Mol Med 24: 129-143.
- Balfour DJ (2015) The role of mesoaccumbens dopamine in nicotine dependence. Curr Top Behav Neurosci 24: 55-98.
- Volkow ND, Koob G, Baler R (2015) Biomarkers in substance use disorders. ACS Chem Neurosci 6: 522-525.
- Bruijnzeel AW (2012) Tobacco addiction and the dysregulation of brain stress systems. Neurosci Biobehav Rev 36: 1418-1441.
- Frost-Pineda K, Muhammad-Kah R, Rimmer L, Liang Q (2014) Predictors, indicators, and validated measures of dependence in menthol smokers. J Addict Dis 33: 94-113.
- Herman AI, DeVito EE, Jensen KP, Sofuoglu M (2014) Pharmacogenetics of nicotine addiction: Role of dopamine. Pharmacogenomics 15: 221-234.
- Yuchuan H, Jie Z, Dongliang L, Ya D, Changguo W, et al. (2011) Circulating biomarkers of hazard effects from cigarette smoking. Toxicol Ind Health 27: 531-535.
- Bierut LJ, Tyndale RF (2018) Preparing the way: Exploiting genomic medicine to stop smoking. Trends Mol Med 24: 187-196.
- Tanner JA, Chenoweth MJ, Tyndale RF (2015) Pharmacogenetics of nicotine and associated smoking behaviors. Curr Top Behav Neurosci 23: 37-86.
- Yu C, McClellan J (2016) Genetics of Substance use disorders. Child Adolesc Psychiatr Clin N Am 25: 377-385.
- Korpi ER, den Hollander B, Farooq U, Vashchinkina E, Rajkumar R, et al. (2015) Mechanisms of action and persistent neuroplasticity by drugs of abuse. Pharmacol Rev 67: 872-1004.
- Jatlow P, Toll BA, Leary V, Krishnan-Sarin S, O’Malley SS (2008) Comparison of expired carbon monoxide and plasma cotinine as markers of cigarette abstinence. Drug Alcohol Depend 98: 203-209.
- Jao NC, Veluz-Wilkins AK, Smith MJ, Carroll AJ, Blazekovic S, et al. (2017) Does menthol cigarette use moderate the effect of nicotine metabolism on short-term smoking cessation? Exp Clin Psychopharmacol 25: 216.
- Van Overmeire IP, De Smedt T, Dendale P, Nackaerts K, Vanacker H, et al. (2016) Nicotine dependence and urinary nicotine, cotinine and hydroxycotinine levels in daily smokers. Nicotine Tob Res 18: 1813-1819.
- Gerra G, Manfredini M, Somaini L, Milano G, Ciccocioppo R, et al. (2016) Perceived parental care during childhood, ACTH, cortisol and nicotine dependence in the adult. Psychiatry Res 245: 458-465.
- Sinha R (2011) New findings on biological factors predicting addiction relapse vulnerability. Curr Psychiatry Rep 13: 398-405.
- Vargas HO, Nunes SO, de Castro MR, Vargas MM, Barbosa DS, et al. (2013) Oxidative stress and inflammatory markers are associated with depression and nicotine dependence. Neurosci Lett 544: 136-140.
- Dempsey D, Tutka P, Jacob P 3rd, Allen F, Schoedel K, et al. (2004) Nicotine metabolite ratio as an index of cytochrome P450 2A6 metabolic activity. Clin Pharmacol Ther 76: 64-72.
- Carim-Todd L, Mitchell SH, Oken BS (2016) Impulsivity and Stress response in nondependent smokers (Tobacco Chippers) in comparison to heavy smokers and nonsmokers. Nicotine Tob Res 18: 547-556.
- McPherson S, Packer RR, Cameron JM, Howell DN, Roll JM (2014) Biochemical marker of use is a better predictor of outcomes than self-report metrics in a contingency management smoking cessation analog study. Am J Addict 23: 15-20.
- Jung HS, Kim Y, Son J, Jeon YJ, Seo HG, et al. (2012) Can urinary cotinine predict nicotine dependence level in smokers? Asian Pac J Cancer Prev 13: 5483-5488.
- Krishnan-Sarin S, Rosen MI, O'Malley SS (1999) Naloxone challenge in smokers. Preliminary evidence of an opioid component in nicotine dependence. Arch Gen Psychiatry 56: 663-668.
- Mushtaq N, Beebe LA, Vesely SK (2012) Determinants of salivary cotinine concentrations among smokeless tobacco users. Nicotine Tob Res 14: 1229-1234.
- Pacek LR, Vandrey R, Dermody SS, Denlinger-Apte RL, Lemieux A, et al. (2016) Evaluation of a reduced nicotine product standard: Moderating effects of and impact on cannabis use. Drug Alcohol Depend 167: 228-232.
- Kaufmann A, Hitsman B, Goelz PM, Veluz-Wilkins A, Blazekovic S, et al. (2015) Rate of nicotine metabolism and smoking cessation outcomes in a community-based sample of treatment-seeking smokers. Addict Behav 51: 93-99.
- Fu M, Martinez-Sanchez JM, Agudo A, Pascual JA, Ariza C, et al. (2012) Nicotine depedence and salivary cotinine concentration in daily smokers. Eur J Cancer Prev 21: 96-102.
- Kwok TC, Taggar J, Cooper S, Lewis S, Coleman T (2014) Nicotine dependence and biochemical exposure measures in the second trimester of pregnancy. Nicotine Tob Res 16: 145-154.
- Benowitz NL, Dains KM, Hall SM, Stewart S, Wilson M, et al. (2009) Progressive commercial cigarette yield reduction: biochemical exposure and behavioral assessment. Cancer Epidemiol Biomarkers Prev 18: 876-883.
- St Helen G, Dempsey D, Wilson M, Jacob P 3rd, Benowitz NL (2013) Racial differences in the relationship between tobacco dependence and nicotine and carcinogen exposure. Addiction 108: 607-617.
- Hatsukami DK, Kotlyar M, Hertsgaard LA, Zhang Y, Carmella SG, et al. (2010) Reduced nicotine content cigarettes: effects on toxicant exposure, dependence and cessation. Addiction 105: 343-355.
- Kawada T, Hirata K, Inagaki H, Otsuka T, Katsumata M (2010) Significance of the 100-point scale to evaluate perceived tobacco dependence. Work 35: 183-189.
- Ferketich AK, Wee AG, Shultz J, Wewers ME (2007) Smokeless tobacco use and salivary cotinine concentration. Addict Behav 32: 2953-2962.
- Muhammad-Kah RS, Hayden AD, Liang Q, Frost-Pineda K, Sarkar M (2011) The relationship between nicotine dependence scores and biomarkers of exposure in adult cigarette smokers. Regul Toxicol Pharmacol 60: 79-83.
- Komiyama M, Wada H, Ura S, Yamakage H, Satoh-Asahara N, et al. (2013) Analysis of factors that determine weight gain during smoking cessation therapy. PLoS One 8: 72010.
- Nunes SO, Piccoli de Melo LG, Pizzo de Castro MR, Barbosa DS, Vargas HO, et al. (2015) Atherogenic index of plasma and atherogenic coefficient are increased in major depression and bipolar disorder, especially when comorbid with tobacco use disorder. J Affect Disord 172: 55-62.
- Cao XZ, Luo ZR, Zhang HM, Cai JQ (2014) Effect of atorvastatin on vascular endothelial function in moderately nicotine-dependent smokers. Genet Mol Res 13: 2698-2702.
- Bortolasci CC, Vargas HO, Vargas Nunes SO, de Melo LG, de Castro MR, et al. (2015) Factors influencing insulin resistance in relation to atherogenicity in mood disorders, the metabolic syndrome and tobacco use disorder. J Affect Disord 179: 148-155.
- Suhaimi MZ, Sanip Z, Jan HJ, Yusoff HM (2016) Leptin and calorie intake among different nicotine dependent groups. Ann Saudi Med 36: 404-408.
- Zaparoli JX, Sugawara EK, de Souza AA, Tufik S, Galduroz JC (2016) Omega-3 levels and nicotine dependence: A cross-sectional study and clinical trial. Eur Addict Res 22: 153-162.
- Korhonen T, Goodwin A, Miesmaa P, Dupuis EA, Kinnunen T (2011) Smoking cessation program with exercise improves cardiovascular disease biomarkers in sedentary women. J Womens Health (Larchmt) 20: 1051-1064.
- Nordskog BK, Brown BG, Marano KM, Campell LR, Jones BA, et al. (2015) Study of cardiovascular disease biomarkers among tobacco consumers, part 2: Biomarkers of biological effect. Inhal Toxicol 27: 157-166.
- Scaglia N, Chatkin J, Chapman KR, Ferreira I, Wagner M, et al. (2016) The relationship between omega-3 and smoking habit: A cross-sectional study. Lipids Health Dis 15: 61.
- Selya AS, Hesse ND (2017) Time to first cigarette and serum cholesterol levels. Soc Sci Med 174: 213-219.
- Rasmusson AM, Wu R, Paliwal P, Anderson GM, Krishnan-Sarin S (2006) A decrease in the plasma DHEA to cortisol ratio during smoking abstinence may predict relapse: A preliminary study. Psychopharmacology (Berl) 186: 473-480.
- al'Absi M, Carr SB, Bongard S (2007) Anger and psychobiological changes during smoking abstinence and in response to acute stress: prediction of smoking relapse. Int J Psychophysiol 66: 109-115.
- al'Absi M, Wittmers LE, Hatsukami D, Westra R (2008) Blunted opiate modulation of hypothalamic-pituitary-adrenocortical activity in men and women who smoke. Psychosom Med 70: 928-935.
- Lee YS, Joe KH, Sohn IK, Na C, Kee BS, et al. (2005) Changes of smoking behavior and serum adrenocorticotropic hormone, cortisol, prolactin, and endogenous opioids levels in nicotine dependence after naltrexone treatment. Prog Neuropsychopharmacol Biol Psychiatry 29: 639-647.
- Buchmann AF, Laucht M, Schmid B, Wiedemann K, Mann K, et al. (2010) Cigarette craving increases after a psychosocial stress test and is related to cortisol stress response but not to dependence scores in daily smokers. J Psychopharmacol 24: 247-255.
- Pomerleau OF, Pomerleau CS, Snedecor SM, Gaulrapp S, Brouwer RN, et al. (2004) Depression, smoking abstinence and HPA function in women smokers. Hum Psychopharmacol 19: 467-476.
- Wardle MC, Munafò MR, de Wit H (2011) Effect of social stress during acute nicotine abstinence. Psychopharmacology (Berl) 218: 39-48.
- Mendelson JH, Goletiani N, Sholar MB, Siegel AJ, Mello NK (2008) Effects of smoking successive low- and high-nicotine cigarettes on hypothalamic-pituitary-adrenal axis hormones and mood in men. Neuropsychopharmacology 33: 749-760.
- Puddey IB, Vandongen R, Beilin LJ, English D (1984) Haemodynamic and neuroendocrine consequences of stopping smoking--a controlled study. Clin Exp Pharmacol Physiol 11: 423-426.
- Krishnan-Sarin S, Rosen MI, O'Malley SS (1999) Naloxone challenge in smokers. Preliminary evidence of an opioid component in nicotine dependence. Arch Gen Psychiatry 56: 663-668.
- von der Goltz C, Koopmann A, Dinter C, Richter A, Rockenbach C, et al. (2010) Orexin and leptin are associated with nicotine craving: A link between smoking, appetite and reward. Psychoneuroendocrinology 35: 570-577.
- Hamidovic A, Khafaja M, Brandon V, Anderson J, Ray G, et al. (2017) Reduction of smoking urges with intranasal insulin: A randomized, crossover, placebo-controlled clinical trial. Mol Psychiatry 22: 1413-1421.
- Cohen LM, al'Absi M, Collins FL (2004) Salivary cortisol concentrations are associated with acute nicotine withdrawal. Addict Behav 29: 1673-1678.
- Morris MC, Mielock AS, Rao U (2016) Salivary stress biomarkers of recent nicotine use and dependence. Am J Drug Alcohol Abuse 42: 640-648.
- Hogle JM, Curtin JJ (2006) Sex differences in negative affective response during nicotine withdrawal. Psychophysiology 43: 344-356.
- Steptoe A, Ussher M (2006) Smoking, cortisol and nicotine. Int J Psychophysiol 59: 228-235.
- al'Absi M, Nakajima M, Grabowski J (2013) Stress response dysregulation and stress-induced analgesia in nicotine dependent men and women. Biol Psychol 93: 1-8.
- Krause D, Warnecke M, Schuetz CG, Soyka M, Manz KM, et al. (2018) The impact of the opioid antagonist naloxone on experimentally induced craving in nicotine-dependent individuals. Eur Addict Res 24: 255-265.
- Umene-Nakano W, Yoshimura R, Yoshii C, Hayashi K, Ikenouchi-Sugita A, et al. (2012) Plasma levels of metabolites of catecholamine in nicotine-dependent patients treated with varenicline. Nicotine Tob Res 14: 486-489.
- Waszkiewicz N, Szajda SD, Waszkiewicz M, Wojtulewska-Supron A, Szulc A, et al. (2013) The activity of serum beta-galactosidase in colon cancer patients with a history of alcohol and nicotine dependence: Preliminary data. Postepy Hig Med Dosw (Online) 67: 896-900.
- Knobloch K, Yoon U, Vogt PM (2011) Preferred reporting items for systematic reviews and meta-analyses (PRISMA) statement and publication bias. J Craniomaxillofac Surg 39: 91-92.
- Van Overmeire IP, De Smedt T, Dendale P, Nackaerts K, Vanacker H, et al. (2016) Nicotine dependence and urinary nicotine, cotinine and hydroxycotinine levels in daily smokers. Nicotine Tob Res 18: 1813-1819.
- Tweed JO, Hsia SH, Lutfy K, Friedman TC (2012) The endocrine effects of nicotine and cigarette smoke. Trends Endocrinol Metab 23: 334-342.
- Rohleder N, Wolf JM, Maldonado EF, Kirschbaum C (2006) The psychosocial stress-induced increase in salivary alpha-amylase is independent of saliva flow rate. Psychophysiology 43: 645-652.
- van Stegeren A, Rohleder N, Everaerd W, Wolf OT (2006) Salivary alpha amylase as marker for adrenergic activity during stress: Effect of betablockade. Psychoneuroendocrinology 31: 137-141.
- Gerlo S, Davis JRE, Mager DL, Kooijman R (2006) Prolactin in man: A tale of two promoters. Bioessays 28: 1051-1055.
- Seyler LE, Fertig J, Pomerleau O, Hunt D, Parker K (1984) The effects of smoking on ACTH and cortisol secretion. Life Sci 34: 57-65.
- Allen AP, Kennedy PJ, Dockray S, Cryan JF, Dinan TG, et al. (2016) The trier social stress test: Principles and practice. Neurobiol Stress 6:113-126.
- Markopoulou K, Papadopoulos A, Juruena MF, Poon L, Pariante CM, et al. (2009) The ratio of cortisol/DHEA in treatment resistant depression. Psychoneuroendocrinology 34: 19-26.
- National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health (2014) The Health Consequences of Smoking-50 Years of Progress : A Report of the Surgeon General. National Center for Chronic Disease Prevention and Health Promotion (US) Office on Smoking and Health, Atlanta, USA.
- Messner B, Bernhard D (2014) Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arterioscler Thromb Vasc Biol 34: 509-515.
- Dierker L, Hedeker D, Rose J, Selya A, Mermelstein R (2015) Early emerging nicotine dependence symptoms in adolescence predict daily smoking in young adulthood. Drug Alcohol Depend 151: 267-271.
- Goldstein BI, Carnethon MR, Matthews KA, McIntyre RS, Miller GE, et al. (2015) Major depressive disorder and bipolar disorder predispose youth to accelerated atherosclerosis and early cardiovascular disease: A scientific statement from the American heart association. Circulation 132: 965-986.
- Bowen KJ, Harris WS, Kris-Etherton PM (2016) Omega-3 fatty acids and cardiovascular disease: Are there benefits? Curr Treat Options Cardiovasc Med 18: 69.
- Dobiášová M (2004) Atherogenic index of plasma [Log(Triglycerides/HDL-Cholesterol)]: Theoretical and practical implications. Clin Chem 50: 1113-1115.
- Nimmanapalli HD, Kasi AD, Devapatla Pk, Nuttakki V (2016) Lipid ratios, atherogenic coefficient and atherogenic index of plasma as parameters in assessing cardiovascular risk in type 2 diabetes mellitus. International Journal of Research in Medical Sciences 4.
- Tonstad S (2009) Cigarette smoking, smoking cessation, and diabetes. Diabetes Res Clin Pract 85: 4-13.
Citation: Newton DF (2019) Peripheral Biomarkers of Tobacco-Use Disorder: A Systematic Review. J Addict Addictv Disord 6: 26.
Copyright: © 2019 Dwight F Newton, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.