PKM2-Mediated Glycolysis and Redox Equilibrium in Cancer Stem Cells: The Metabostemness Phenomenon
*Corresponding Author(s):
Stavroula D ManolakouDepartment Of Medical Oncology, University Hospital Of Heraklion, Heraklion, Crete, Greece
Tel:+03 06976857042,
Email:sdmanolakou@hotmail.com
Abstract
The discovery of novel targets in cancer cells along with the rapid development of innovative therapeutic agents against them and especially in last decade has led to better cancer survival rate. However, the existence of Cancer Stem Cells (CSCs) into the tumor colonies has been suggested as a crucial impediment in cancer fight. Not only the self-renewal and high proliferative ability of CSCs but also the diversity of their metabolic signatures amplifies the tumor heterogeneity and treatment-resistance, defining a new hallmark of oncogenesis called metabostemness phenomenon. This phenomenon concludes several patterns of metabolic behaviour which rely on three basic phenotypes; (i) Aerobic glycolysis or Warburg effect; (ii) Reverse Warburg effect and (iii) Oxidative Phosphorylation (OXPHOS) phenotype. Pyruvate Kinase M2 (PKM2) has been considered as the key regulator enzyme of metabolic reprogramming of CSCs as alternates itself between a dimer state, responsible for aerobic glycolysis and a tetramer state, responsible for OXPHOS procedure. Several PKM2 interactions which perceive the changes of a usually oxidative microenvironment of CSCs attribute to a metabolic plasticity and as a result to survival, proliferation, metastasis and drug-resistance. Henceforth, this review highlights the evidence regarding CSCs about these PKM2-mediated interactions describing the outcome of them in terms of metabolic reprogramming and of maintenance of redox equilibrium. Thereafter, a recapitulation of molecular targeting of PKM2 and metabostemness will be attempted so as to be given prominence to the development of innovative metabolic targets and therapies against the triangle “PKM2 - metabolic plasticity - oxidative scale” in CSCs.
Keywords
Aerobic glycolysis; Cancer stem cells; Oxidative stress; Metabostemness; PKM2; Stemness
INTRODUCTION
Last twenty years, several anticancer therapies have been developed and multiple treatment strategies against tumor cells have been suggested in order serious malignancies to be faced. Indeed, according to recent epidemiological data in past decade the cancer death rate seems to be declined [1]. Nevertheless, multiple molecular “escape” mechanisms against therapies have been demonstrated with a particular reference to inhibition of apoptosis’ pathways, induction of drug detoxification proteins, alteration of nucleic acid damage repair system and activation of several survival mechanisms [2]. Among these survival signals that contribute in treatment-resistance is the phenomenon of “cancer stemness” [2,3]. Generally, the term “stemness” has been defined as the ability of Embryonal (ESCs) or Pluripotent Somatic Cells (PSCs) to self-renew perpetuating their lineage and to interact with their microenvironment giving the rise to differentiated cells. Moreover, an appropriate stimuli i.e., epigenetic event is able to induce vice versa the reprogramming of progenies to dedifferentiated status, the “stem-cell” status [4].
However, when somatic cells and normal somatic Stem Cells (SCs), or even precancerous adult cells exploit their pluripotency to sustain survival advantage even into toxic microenvironment without maintaining tissue homeostasis, the progeny clones can be transformed into resistant Cancer Stem Cells (CSCs) with aggressive molecular characteristics [4,5]. Specifically, mainly four crucial traits differentiate the phenomenon of “stemness” from “cancer stemness” [4]; abnormal activation of self-renewal pathways such as Notch, Hedgehog, PTEN, TGF-β or Wnt, short cell cycles with attenuated DNA repair mechanisms, induced cell plasticity (i.e., epithelial-mesenchymal transition) and conversion of cell metabolic status from oxidative and energy-producer phenotype to glycolytic and biosynthetic phenotype or vice versa [6,7].
The latter phenomenon regarding metabolic remodeling in cancer was first described by Otto Warburg in 1920s [8,9]. This observation is known as Warburg effect and was defined as the preference of cancer cells to perform aerobic glycolysis rather than Oxidative Phosphorylation (OXPHOS) even in normoxic conditions, increasing their biosynthetic properties but with detriment to ATP production [10-12]. Recent studies have shown that ESCs and CSCs respond rapidly in their high anabolic needs via aerobic glycolysis activating biosynthetic pathways like Pentose Phosphate Pathway (PPP) and in turn sustain their self-renewal ability and pluripotency [7,13,14]. However, the hyper-energetic activity of SCs induces the production of Reactive Oxygen and Nitrogen Species (ROS and RNS) increasing the intracellular oxidative load. Epigenetic interactions between glycolytic pathways and antioxidant defense pathways such as Hypoxia-Inducible Factor 1-alpha (HIF1α), Nuclear Factor κappa B (NFκB) and nuclear factor (erythroid-derived 2)-like-2 (Nrf2) have been described in CSCs as a survival hijacking that lead in retrieval of intracellular redox balance [15-18].
In this review, we firstly describe the metabolic plasticity of CSCs recapitulating the interfering molecular pathways and in the second step we focus on Pyruvate Hinase Isoform M2 (PKM2) which is the key regulator of aerobic glycolysis shedding light on its redox metabolic regulatory action. In this way we highlight crucial molecular players that may be exploited in future innovative therapeutic strategies.
METABOLIC PLASTICITY IN CSCS: THE METABOSTEMNESS PHENOMENON
Since 1970s, especially after the elucidation of impact of Warburg effect on carcinogenesis, metabolic modifications in cancer cells had been considered as a novel hallmark in cancer and had been mainly pertained to the increased uptake of glucose from cancer cells along with induced anabolic glycolytic processes [9]. However, to explain the tumor aggressiveness and heterogeneity several studies additionally investigated the CSC model of carcinogenesis where a distinct highly tumorigenic population of SCs would have an asymmentric division to birth identical SCs but in parallel to produce more differentiated progenies [19,20]. This stemness phenomenon in cancer in combination with the distinct metabolic phenotypes of CSCs even into the same SC niche or tumor colony configured a new hallmark concept called metabostemness. Reversely, the metabostemness theory reflect the metabolic plasticity in different types of CSCs and of cancer cells with their metabolic status ranged from dormant to aggressive, from hyperoxic or normoxic to hypoxic and from oxidative to glycolytic [21]. Indeed, different oxygenation levels or other stimuli such as inflammatory signals, irradiation, malnutrition or treatment-induced stress can activate or inactivate certain metabolic epigenetic signatures or even more reprogram specific cancer metabotypes in favour or not of CSC-like phenotype [19].
CSC model of carcinogenesis firstly identified in acute myeloid leukaemia, when Lapidot et al., and Bonnet et al., discovered that a CD34+/CD38- cell population developed in a hierarchical manner into immunodeficient mice [22,23]. Since then, a cataract of studies revealed in general terms that mainly three metabolic and bioenergetic phenotypes can be recognized in CSCs and can be altered even into the same tumor population serving in complexity and heterogeneity of cancer; (i) Warburg effect or Glycolytic phenotype, (ii) Reversed Warburg phenotype, and (iii) OXPHOS phenotype [24].
For instance, a metabolic switch in favor of anabolic pathways such as PPP and against OXPHOS has been detected in in vitro and in vivo experiments in brain CSCs, in glioma cells and in human glioblastoma tissues [25]. In particular, these cells exploit the biosynthetic advantage of Warburg effect and acquire high proliferating dynamic. For this reason, elevated telomerase expression and long leukocyte telomere length have been revealed in gliomagenesis and glioma cells [26]. Similarly, CD44+/CD24-/EPCAM+ breast stem-like cells maintain their stemness via increased glucose uptake and aerobic glycolysis [27].
However, the “seed and soil” theory states that an appropriate tumor microenvironment is a prerequisite for a tumor SC niche development and proliferation. Based on this theory, the reverse Warburg phenotype has been described. In this phenotype CSCs interact with each-other and with cancer-associated fibroblasts (CAFs) in a way that lactate, pyruvate, amino acids and ketone bodies derived from CAFs to perform mitochondrial respiration are catabolised and on the other hand the anabolic products from CAFs via aerobic glycolysis are elicited [28,29]. Indeed, it is well-reported that CAFs can facilitate the reprogramming of cancer cells into CSCs and enhance the CSCs’ resistance via excretion of pro-stemness chemokines and cytokines i.e., NF-κB, IL6, IL8, prostaglandins [30]. However, monocarboxylate transporters (MCTs) seem to have a central role in terms of CSCs-CAFs metabolic symbiosis as glycolytic CAFs export lactate via MCT4 and oxidative cancer cells or CSCs import lactate via MCT1 [31]. For instance, in triple negative breast cancer tissues high expression of Monocarboxylate Transporter (MCT4) biomarker of aerobic glycolysis has been detected only in stromal cells, a fact that is validated in radiation-resistant breast CSCs where increase of mitochondrial population and OXPHOS prominence were detected in contrast to neighbouring breast differentiated progeny cells [32,33]. Similarly, in glioblastoma cells only periastrocytes are responsible for cAMP-CREB-PGC1α pathway activation inducing mitochondrial respiration and differentiation of glioblastoma cells [34].
Regarding OXPHOS phenotype, it has been demonstrated that somatic SCs can present this metabolic behaviour to acquire high energy despite high levels of ROS are concurrently produced. When SCs enhance their pluripotency and their self-renewal, the risk of oxidative damage rises and either a reverse Warburg effect neutralize the oxidative load or a metabolic switch to glycolytic phenotype is performed [35]. This metabolic plasticity has been demonstrated in Lgr5+ intestinal SC niche baring the OXPHOS phenotype of Lgr5+ SCs and the glycolytic phenotype of Paneth cells [36]. Another paradigm is glioblastoma CSCs that mainly exploit the OXPHOS to enhance their energy needs. To be more specific, in in vitro glioblastoma CSCs, Insulin Growth Factor 2 Binding Protein 2 (IGF2BP2) was overexpressed leading to regulation of mitochondrial respiratory chain complex and to OXPHOS metabolic phenotype. Importantly, knock-down of IGF2BP2 not only affected OXPHOS rate but also reduced glioblastoma “stemness” and tumorigenity [37]. Moreover, leukemic cells mainly appear OXPHOS phenotype with increase of intracellular oxidative load but with parallel increase of antioxidant genes’ expression [38]. Indeed, tigecycline, an antibiotic targeting mitochondrial gene expression presents an inhibiting activity against chronic myeloid leukemic CSCs ‘proliferation inducing chemotherapy sensitivity [39].
Several studies have already recognised multiple metabolic regulators that can induce the switch from the one metabolic phenotype to another. NANOG, Oct4, SOX-2, KLF-4 transcription factors along with cMYC multivalent protein (OSKM-N complex) can induce the pluripotency of SCs and their conversion to CSCs, a fact that leads in higher oxidative status [6,40-42]. However, the mitochondrial inhibition, mitophagy and mitochondrial fission via KLF-4, SOX-2 and cMYC transcriptional activity promote the Warburg effect and the protection of CSCs from hyper-oxidative stress [7,43]. Additionally, overexpression of cMYC has been correlated with high PKM2/PKM1 status on CSCs indicating SCs reprogramming into glycolytic phase and concurrently facilitating their survival expansion [44]. Except for transcriptional factors, several oncogenes, tumor suppressor genes and membrane proteins have been studied for their role in metabolic plasticity. For instance, CAIX transmembrane protein can induce EMT phenotype and stemness via Notch and Jagged pathways in breast CSCs and CD44v membrane glycoprotein indirectly facilitates GSH synthesis reducing accumulating ROS a fact that in turn induce glycolysis but not anabolic PPP. Interestingly, MYC low/CD44 high CSC phenotype has been shown as a dormant metabolic phenotype, a result which has been confirmed by the observation that a component of ubiquitin ligase, known as FBW7, resulting in proteasome - dependent degradation of cMYC facilitate the quiescent status [45]. Moreover, in hepatocellular carcinoma loss of p62 has been associated with lower glucose uptake, lower glutamine metabolism and decreased PPP [14]. However, the abundance of the metabolic regulators and the complex interactions between them reflect the need for more studies to decipher the metabolic reprogramming of CSCs.
PKM2: THE CENTRAL ENZYME IN CSCS’ GLYCOLYSIS
PKM2 is a glycolytic enzyme involved in the final but pace-making step of glycolysis and is expressed in adult SCs, in CSCs and in cancer cells. In particular this enzyme catalyses the transfer of phosphate from phosphoenolpyruvate (PEP) to Adenosine 5’ - Dihosphate (ADP) so that pyruvate and Adenosine 5’ - Triphosphate (ATP) are synthesized. In mammals, four isoforms of Pyruvate Kinase (PK) have been described; PKL, PKR, PKM1, and PKM2 [46]. However, in SCs and in carcinogenesis, the expression of PKM2 becomes gradually dominant over PKL/ R/ M1 expression [47]. Notably, it remains unclear whether PKM1 undergoes conversion in PKM2 in cancer cells and in CSCs or if the alternative splicing of PKM gene is alternated during tumorigenesis.
It has been already demonstrated that PKM2 undergoes an allosteric regulation that refers to switch between a tetrameric and a dimeric state (R/T state). Particularly, on the one hand the tetrameric PKM2 has high catalytic activity and is involved in catabolic processes leading to ATP synthesis, whereas the dimeric PKM2 has been characterised by its low catalytic activity with inductive action on cell glycolytic pathways (i.e., PPP) [46,48]. Several studies have shown that tumor cells activate the dimeric state of PKM2 to achieve anabolic demands of oncogenesis. Fructose-1,6-biphosphate controls as an allosteric activator the switch of PKM2 from dimer to tetramer form. Nevertheless, the presence of intermediate anabolic metabolites such as serine and Succinyl-Amino-Imidazole Carboxamide-Ribose-5′-phosphate (SAICAR) function as negative feedback for synthetic dimer PKM2 inducing its tetramerization [48-51]. In contrast, PKM2 acetylation (i.e., 305 Lys), PKM2 OGlcNacylation and PKM2 phosphorylation (i.e., Y105) can be stimulated by high glucose and amino acids (i.e., alanine, phenylalanine) concentrations and by hypoxic conditions resulting in inhibition of enzymatic activity of PKM2 and stabilisation of PKM2 dimer state [10,51,52]. Moreover the acetylation of PKM2 facilitates the interaction between PKM2 and autophagy charepones leading PKM2 to lyposomic degradation [53]. Notably, in carcinogenesis dimer PKM2 has also been characterised as a non-metabolic transcriptional co-activator enhancing glycolytic phenotype and protecting CSCs from high oxidative load and hostile microenvironment [11,54-56]. For instance, PKM2 transcriptionally regulates the expression of CCND1, cMYC, GLUT1 and LDHA proteins via activation of Wnt/ β-catenin pathway. In this way, PKM2 seems to orchestrate tumor cell proliferation, glucose uptake and aerobic glycolysis. Despite the transcriptional role of PKM2, several cross-talks between PKM2 and oncogenic pathways (i.e steroid sulfatase, NF-κB) can induce angiogenesis, EMT and metastasis [10,57].
Importantly, the role of PKM2 in oncogenesis and in stemness has been investigated for various CSCs types. PKM2 expression enhances prostate CSCs’ proliferation in experiments with DU145 prostate CSCs and this is in agreement with the higher levels of PKM2 expression in Gleason 8-10 score prostate tissues in contrast to Gleason 6-7 score tissues. In addition, PKM2 expression was enhanced in Pancreatic Ductal Adenocarcinoma (PDAC) tissues and cell lines and more importantly on PKM2 silencing the percentage of PDAC cells that entered into G0/G1 phase was impressively elevated. The latter indicates a crucial intervening role of PKM2 in cell cycle and renewal effect. However PKM2 affects cell cycle not only via the regulation of cyclin-dependent kinase, but also via alteration of CSC phenotype. Indeed, in in vitro experiments on A549 - derived CSCs the knockdown of PKM2 resulted on reduction of CD44 marker and on elimination of cells’ capacity to form spheroids. In concordance, in a study derived from our group, in ovarian cancer tissues, PKM2low/CD44low patients had better prognosis compared to PKM2high/ CD44high patients [58]. Another activity of PKM2, as also described previously, is its interaction with Oct4 protein regulating CSCs’ death and differentiation. Experiments both in P19 embryonic carcinoma cell extraction and in glioma SCs proved through PKM2 silencing techniques that PKM2 is involved on CSCs spheroid differentiation via binding to POU domain of Oct-4. Moreover, studies in liver CSCs have focused on mi-RNAs (miRs) revealed that miR-675 in cooperation with PKM2 leads to formation of the appropriate activator complex that binds to cMYC promoter so as to induce its transcription. Similarly, liver CSCs proliferation can be enhanced by miR-122 that importantly attenuates PKM2 expression. Ηοwever, the limpid net of PKM2 interactions that promotes glycolytic phenotype in CSCs and induces stemness has not been yet fully clear.
PKM2: A CENTRAL REGULATOR OF CSCS’ REDOX EQUILIBRIUM
Stress is experienced by cells when pro-oxidant and electrophilic reactive species (i.e., ROS and RNS) overwhelm the cell’s antioxidant and detoxification proteins. In addition to causing protein and lipid damage, stress products can cause mutations and epigenetic perturbation by damaging DNA and proteins that modify chromatin. Ischemia, hypoxia or hyperoxia, heat, radiation and toxins could be some of the exogenous stress factors that disrupt intracellular redox equilibrium [59]. However, depending on endogenous stress factors, several types of stress have been proposed including apoptotic, oxidative, metabolic, mitotic, proteostatic and hypoxic stress. Cancer cells and especially CSCs that exhibit self-renewal and rapid proliferative properties appear disturbance of redox balance with ROS and RNS to be continuously increased. For this reason, cancer cells and CSCs secondarily activate survival or/and antiapoptotic pathways to hijack hyper-stress conditions. In particular, oxidative free radicals can induce calcium/calmodulin pathway releasing calcium from intracellular deposits and finally activating kinases (i.e., Protein Kinase C (PKC)) that promote cell survival, cytoskeleton remodelling and cell motility [60]. ERK1/2 and PI3K/Akt pathways assume to be activated after increasing of oxidative load as well as several antioxidant transcription factors or proteins.
Notwithstanding, metabolic plasticity of CSCs and the switch to glycolytic phenotype mediated by PKM2 have been proposed as crucial events for the maintenance of redox equilibrium. Even more, Movahed et al., suggest that the deal of cancer cells to overcome oxidative stress acquiring Warburg effect phenotype may be the crucial step of conversion from cancer cells to CSCs [14]. Indeed, studies with hepatocellular carcinoma cells in oxidative stress revealed that PKM2 converts into dimer state and enter into mitochondria where interact with apoptotic Bcl2. The PKM2-mediated phosphorylation of Bcl2 inhibits the proteasomal-degradation of Bcl-2 leading to anti-apoptotic signalling and cancer cell survival [61]. In gastric carcinoma, PKM2 seems to enhance “cancer stemness” via stabilisation of p65 subunit of NFκΒ. In this way, NFκB interact with and in turn activate Bcl-xL antiapoptotic protein promoting gastric cancer development [62].
Multiple experiments in breast, prostate, bladder and brain cancer cells demonstrate that hypoxic stress can activate HIF-1α transcription factor mediating the transcriptions of glycolytic enzymes such as PKM2 and glucose transporters, GLUT1 and CAIX [14,25,63,64]. Strict regulation of HIF-1α has been demonstrated as in low ROS levels HIF-1α is activated and in turn induces glycolysis, whereas in high ROS levels it is attenuated and thus, PKM2 dimer state and PPP pathway are induced [14].
All the above observations reflect the ability of oxidative free radicals to act as mediators on PKM2 allosteric conversion and subsequently on metabolic reprogramming on CSCs. However, the molecular mechanisms, with which PKM2 maintain CSCs redox equilibrium, is still under investigation. It is well - known that dimer state of PKM2 has low catalytic power and as a result there is great accumulation of upstream products of glycolysis pathway that in turn enter into anabolic pathways. So, glucose-6-phosphate is leaded to PPP pathway where it turns into ribose-5-phosphate. During these reactions Glucose-6-Phosphate Deydrogenase (G6PD) is the determining enzyme that uses NADP+ as a cofactor which in turn neutralise free radicals via NADPH formation. In other words, PKM2 dimer state can recover redox equilibrium increasing NADPH/NADP ratio [65]. Another mechanism of PKM2-driven ROS reduction is the Mitochondrial Membrane Potential (MMP). In bone marrow mesenchymal SCs’ experiments, PKM2 inhibitor (C3k) resulted in increase of MMP and a reduction of oxidative stress was observed [66]. A complicated mechanistic sequence has been described for liver CSCs where a long-noncoding RNA HULC enhanced the growth of CSCs via miR675-PKM2 autophagy. The latter reduced proteostatic stress and induced cell cycle and proliferation kinases [67]. Another mechanism of how PKM2 affects oxidative status has been demonstrated from studies on Histone Deacetylase (HDAC) inhibitors. In particular, PKM2-mediated PPP metabolism and increase of NADPH/NADP ratio were detected in osteosarcoma cell lines where Aldeyde Dehydrogenase (ALDH+) cells treated with valproic acid (HDAC inhibitor) appear induction of stemness [68]. Finally, the cooperation of PKM2 with Nrf2 - which is the central transcriptional factor of antioxidant and detoxification genes (i.e., ARE genes) - is included on the mechanisms that PKM2 maintain redox equilibrium. Nrf2 has been demonstrated as the central molecule in the Nrf2-Keap-ARE signalling pathway [69] and in basal conditions, it is targeted for proteasomal degradation by its cytoplasmic inhibitor, Kelch-like ECH-associated protein 1 (Keap1), while in oxidative conditions, Nrf2 degradation is abolished and Nrf2 accumulates in the nucleus where it transactivates protective genes [69,70]. Recent studies have shown that Nrf2 induces the transcription not only of antioxidant genes but also of metabolic-related genes such as PKM2 [71]. Μoreover, in vitro experiments have revealed that the Nrf2 pathway also facilitates the glucose uptake and the import into PPP [72-74]. However, the exact implication of Nrf2-PKM2 complex on metabolic plasticity of CSCs and on redox equilibrium remains obscure.
TARGETING OF PKM2: A NOVEL ANTICANCER TREATMENT AGAINST METABOSTEMNESS
Several in vitro and in vivo experiments have demonstrated the use of Small Hairpin (SH) RNAs or chemical compounds that lead to inhibition of PKM2 or stabilization of PKM2 tetramer. This rationale could be used as a therapeutic strategy against metabolic reprogramming of cancer cells and against metabostemness.
Indeed, PKM2 inhibition has been proposed in upregulation of cells’ platinum sensitivity reducing cell proliferation and increasing cell apoptosis [55,75]. However, although silencing of pkm2 gene in HT29 intestinal cells interestingly increased the platinum resistance, in p53 wild type HCT116 intestinal cells pkm2 silencing induced chemosensitivity [76]. Likewise, inhibition of PKM2 via miR-122 in 5-Fluorouracil (5FU) resistant colon cancer cells led to cell resensitization in 5FU [77]. These controversial results reveal that the diversity of CSCs metabolic profiles among several cancer types and among different microenvironments is a hard pitfall for the investigators to discover whether PKM2 targeting would have anticancer effect. Experimental models with xenografts and organoids may recapitulate the features of metabostemness and give information for the appropriate therapeutic strategy.
Data for several types of cancer show that when PKM2 expression is upregulated cancer cells’ or CSCs’ treatment sensitivity is arrested (Table 1). Targeting of PKM2 and metabostemness has been suggested as an innovative rationale for development of anticancer agents. Shikonin is a herbal extract from lithospermum erythrorhizon that has been proposed as a novel PKM2 inhibitor resensitizing T24 bladder cancer cells in cis-platinum treatment [78,79]. In ML526 and MEL697 melanoma cells another novel PKM2 inhibitor has been tested; called lapachol. Lapachol is a natural compound isolated from the tree, handroanthus impetiginosus and seems to sensitize ML526 and MEL697 cells to apoptosis causing ferrotoxicosis and inhibiting glycolysis either in normoxic or hypoxic conditions [80]. This strategy is in concordance with studies that support the metabolic glycolytic phenotype of melanoma cells along with the dysfunction of OXPHOS metabolic machine [81]. Vitamin K (VK) family members belong to naphthoquinones chemical group and several experiments have proposed VK3 and 5 as promising selective PKM2 inhibitors. In immortalised human T-lymphocytes and myelogenous leukaemia cell line, VK3 and Vitamin C improved synergistically the effectiveness of doxorubicin [82,83]. Compound 3k which is a novel synthetic naphthoquinone derivative has also been characterised as selective inhibitor of PKM2 better than shikonin. This anticancer effect of compound 3k revealed in experiments with HCT116, Hela and H1299 cells and by injecting it into xenografts [84]. TEPP-46 and DASA-58 are two selective pyruvate kinase activators meaning that stabilize the PKM2 enzyme in tetramer state. Studies in human cancer cell xenografts suggest that these PKM2 activators can inhibit oncogenesis and alternate cancer metabolism in lung cancer cell cultures [55]. Another antineoplastic PKM2 inhibitor is the synthetic peptide TLN232 which is under evaluation in clinical trials (phase II) for recurrent melanoma [85]. Even more, a stable sesquiterpene lactone called micheliolide accelerating the PKM2 acetylation and PKM2 nuclear translocation inhibit leukaemia and glioma cells to survive and proliferate [86,87]. In terms of repurposing already approved treatments, metformin has been demonstrated to improve platinum-sensitivity of osteosarcoma stem cells downregulating PKM2 levels [88]. Moreover, metformin has shown anticancer activity on gastric cancer cells via HIF1α/PKM2 inhibition as well as on bladder cancer cells treated with docetaxel, trastuzumab and pertuzumab [89,90]. Although benserazide belongs to established and approved anti-Parkinson treatment, studies on BRAFi resistant melanoma cells (SK-MEL5/28) and on human embryonic kidney cells (HEK293) showed benserazide’s inhibitory effect on PKM2 resulting in apoptosis and avoidance of metastasis [91]. Finally, the first molecule (MS-001) against metabolic plasticity of CSCs has been already designed by MetaboStem biotechnology company but its exact activity and effectiveness remain under investigation [92].
|
Agent |
Source |
Chemical category |
Indication |
Studies |
Reference |
|
Shikonin
|
Natural (lithospermum erythrorizon) |
Alkannin |
|
Bladder Cancer |
[79] |
|
Lapachol |
Natural (handroanthus impetiginosus) |
Naphthoquinone |
|
Melanoma cell lines |
[93] |
|
Compound 3k |
Synthetic |
Naphthoquinone |
|
Colon, Cervical, NSCLC cell lines |
[84] |
|
Vitamin K |
Natural |
Fat-soluble vitamin |
|
T-lympoma, Myelogenous leukaemia cell lines |
[82] |
|
Micheliolide |
Natural (Michelia spp.) |
Sesquiterpene lactone |
|
Leukaemia, glioma cell lines |
[87,94] |
|
TEPP-46 |
Synthetic |
Organosulfur heterocyclic, organonitrogen heterocyclic, organic heterobicyclic compound C17H16N4O2S2 |
|
Lung cancer cell lines, Xenografts |
[54] |
|
DASA-58 |
Synthetic |
A member of benzenes and a sulfonamide C19H23N3O6S2 |
|
Lung cancer cell lines, Xenografts |
[54] |
|
TLN-232 |
Synthetic |
Cyclic heptapeptide |
|
Recurrent melanoma (clinical phase II) |
[84] |
|
Metformin |
Synthetic |
Crystalline compound C4H11N5 • HCl |
Treatment of type 2 diabetes |
Breast, bladder, gastric cancer cell lines |
[89,90] |
|
Benserazide |
Synthetic |
Carbohydrazide C10H15N3O5 |
Aromatic L-amino acid decarboxylase or DOPA decarboxylase inhibitor |
BRAFi resistant melanoma cell lines |
[91] |
Table 1: Therapeutic agents against PKM2 and cancer stemness.
CONCLUSION
CSCs have been characterised as the origin of cancer cells and their ability of self-renewal and asymmetric division along with metabolic plasticity conform the phenomenon of metabostemness. This phenomenon has been proposed as a hallmark of chemoresistance, radioresistance, and cancer recurrence or metastasis. The metabolic phenotypes of CSCs alternated each other in a way that CSCs can adapt into specific usually hostile environment conditions. PKM2 seems to be a key regulator of metabolic reprogramming of CSCs perceiving in parallel the disturbance of redox equilibrium. Consequently, a hypothesis of “the clock and the scale” can be created (Figure 1); the metabolic clock of CSCs has a dependent relation with oxidative scale and PKM2 regulates this relation. The exact factors that affect this triangle and especially PKM2 remain clear, although multiple therapeutic strategies against metabostemness have already been underway.
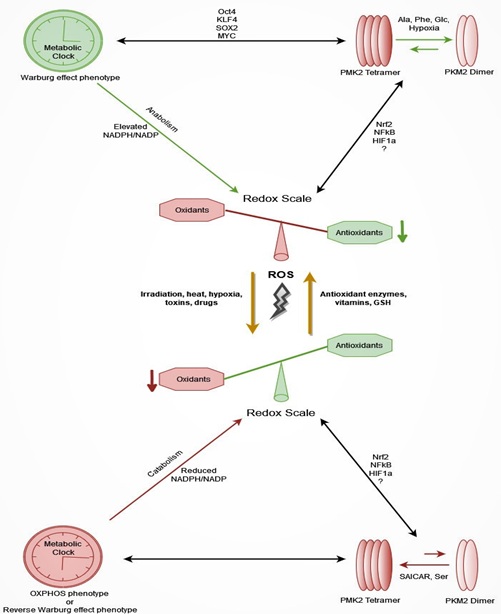
Figure 1: The hypothesis of “the metabolic clock and the redox scale”: CSCs are characterised by metabolic plasticity conforming the phenomenon of metabostemness, a novel hallmark of treatment resistance. Metabolic phenotypes of CSCs; (i) Warburg effect phenotype, (ii) OXPHOS phenotype, and (iii) Reverse Warburg phenotype alternated each other in a way that CSCs can adapt into oxidative environment conditions. PKM2, the pace-making step enzyme of gylycolysis via its allosteric transformation crucially regulates metabolic reprogramming of CSCs perceiving in parallel the disturbance of redox equilibrium. On the one hand (top part of image), the NADPH/NADP ratio is elevated when anabolic procedures (PKM2 dimerism) are triggered, and oxidative stress is reduced. Simultaneously, antioxidant protective enzymes and proteins are overexpressed when transcription factors like Nrf2, NFκB and HIF1α are activated via high oxidative load. On the other hand (bottom part of image), the NADPH/NADP ratio is reduced when catabolic procedures are induced (PKM2 tetramerism), and intracellular oxidative stress is elevated. In these circumstances restoration of intracellular redox balance is restored by activation of antioxidant transcription factors as well as by the reverse Warburg effect on which CSC niche microenvironment participates.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
- Siegel RL, Miller KD, Jemal A (2019) Cancer statistics, 2019. CA Cancer J Clin 69: 7-34.
- Sakthivel KM, Hariharan S (2017) Regulatory players of DNA damage repair mechanisms: Role in Cancer Chemoresistance. Biomed Pharmacother 93: 1238-1245.
- Zhao Z, Song Z, Liao Z, Liu Z, Sun H, et al. (2016) PKM2 promotes stemness of breast cancer cell by through Wnt/beta-catenin pathway. Tumour Biol 37: 4223-4234.
- Aponte PM, Caicedo A (2017) Stemness in Cancer: Stem Cells, Cancer Stem Cells, and Their Microenvironment. Stem Cells Int 2017: 5619472.
- O'Brien CA, Kreso A, Jamieson CH (2010) Cancer stem cells and self-renewal. Clin Cancer Res 16: 3113-3120.
- Spyrou J, Gardner DK, Harvey AJ (2019) Metabolism Is a Key Regulator of Induced Pluripotent Stem Cell Reprogramming. Stem Cells Int 2019: 7360121.
- Nishimura K, Fukuda A, Hisatake K (2019) Mechanisms of the Metabolic Shift during Somatic Cell Reprogramming. Int J Mol Sci 20: 2254.
- Warburg O (1956) [Origin of cancer cells]. Oncologia 9: 75-83.
- Warburg O (1956) On the origin of cancer cells. Science 123: 309-314.
- He X, Du S, Lei T, Li X, Liu Y, et al. (2017) PKM2 in carcinogenesis and oncotherapy. Oncotarget 8: 110656-110670.
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, et al. (2011) Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 334: 1278-1283.
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, et al. (2008) The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230-233.
- Dayem AA, Choi HY, Kim JH, Cho SG (2010) Role of oxidative stress in stem, cancer, and cancer stem cells. Cancers (Basel) 2: 859-884.
- Ghanbari Movahed Z, Rastegari-Pouyani M, Mohammadi MH, Mansouri K (2019) Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed Pharmacother 112: 108690.
- Jang J, Wang Y, Kim HS, Lalli MA, Kosik KS (2014) Nrf2, a regulator of the proteasome, controls self-renewal and pluripotency in human embryonic stem cells. Stem Cells 32: 2616-2625.
- Esteban MA, Wang T, Qin B, Yang J, Qin D, et al. (2010) Vitamin C enhances the generation of mouse and human induced pluripotent stem cells. Cell Stem Cell 6: 71-79.
- Ji J, Sharma V, Qi S, Guarch ME, Zhao P, et al. (2014) Antioxidant supplementation reduces genomic aberrations in human induced pluripotent stem cells. Stem Cell Reports 2: 44-51.
- Hawkins KE, Joy S, Delhove JM, Kotiadis VN, Fernandez E, et al. (2016) NRF2 Orchestrates the Metabolic Shift during Induced Pluripotent Stem Cell Reprogramming. Cell Rep 14: 1883-1891.
- Menendez JA, Alarcon T (2014) Metabostemness: a new cancer hallmark. Front Oncol 4: 262.
- Menendez JA (2015) The Metaboloepigenetic Dimension of Cancer Stem Cells: Evaluating the Market Potential for New Metabostemness-Targeting Oncology Drugs. Curr Pharm Des 21: 3644-3653.
- Menendez JA, Corominas-Faja B, Cuyas E, Alarcon T (2014) Metabostemness: Metaboloepigenetic reprogramming of cancer stem-cell functions. Oncoscience 1: 803-806.
- Bonnet D, Dick JE (1997) Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med 3: 730-737.
- Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, et al. (1994) A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367: 645-648.
- Shen YA, Pan SC, Chu I, Lai RY, Wei YH (2020) Targeting cancer stem cells from a metabolic perspective. Exp Biol Med (Maywood) 245: 465-476.
- Strickland M, Stoll EA (2017) Metabolic Reprogramming in Glioma. Front Cell Dev Biol 5: 43.
- Walsh KM, Codd V, Rice T, Nelson CP, Smirnov IV, et al. (2015) Longer genotypically-estimated leukocyte telomere length is associated with increased adult glioma risk. Oncotarget 6: 42468-42477.
- Dong C, Yuan T, Wu Y, Wang Y, Fan TW, et al. (2013) Loss of FBP1 by Snail-mediated repression provides metabolic advantages in basal-like breast cancer. Cancer Cell 23: 316-331.
- Fu Y, Liu S, Yin S, Niu W, Xiong W, et al. (2017) The reverse Warburg effect is likely to be an Achilles' heel of cancer that can be exploited for cancer therapy. Oncotarget 8: 57813-57825.
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, et al. (2009) The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 8: 3984-4001.
- Chan TS, Shaked Y, Tsai KK (2019) Targeting the Interplay Between Cancer Fibroblasts, Mesenchymal Stem Cells, and Cancer Stem Cells in Desmoplastic Cancers. Front Oncol 9: 688.
- Payen VL, Mina E, Van Hee VF, Porporato PE, Sonveaux P (2020) Monocarboxylate transporters in cancer. Mol Metab 33: 48-66.
- Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, et al. (2012) Using the "reverse Warburg effect" to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle 11: 1108-1117.
- Vlashi E, Lagadec C, Vergnes L, Reue K, Frohnen P, et al. (2014) Metabolic differences in breast cancer stem cells and differentiated progeny. Breast Cancer Res Treat 146: 525-534.
- Xing F, Luan Y, Cai J, Wu S, Mai J, et al. (2017) The Anti-Warburg Effect Elicited by the cAMP-PGC1alpha Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep 18: 468-481.
- Peixoto J, Lima J (2018) Metabolic traits of cancer stem cells. Dis Model Mech 11: 033464.
- Rodriguez-Colman MJ, Schewe M, Meerlo M, Stigter E, Gerrits J, et al. (2017) Interplay between metabolic identities in the intestinal crypt supports stem cell function. Nature 543: 424-427.
- Janiszewska M, Suva ML, Riggi N, Houtkooper RH, Auwerx J, et al. (2012) Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev 26: 1926-1944.
- Khan AUH, Rathore MG, Allende-Vega N, Vo DN, Belkhala S, et al. (2016) Human Leukemic Cells performing Oxidative Phosphorylation (OXPHOS) Generate an Antioxidant Response Independently of Reactive Oxygen Species (ROS) Production. EBioMedicine 3: 43-53.
- Kuntz EM, Baquero P, Michie AM, Dunn K, Tardito S, et al. (2017) Targeting mitochondrial oxidative phosphorylation eradicates therapy-resistant chronic myeloid leukemia stem cells. Nat Med 23: 1234-1240.
- Muller M, Hermann PC, Liebau S, Weidgang C, Seufferlein T, et al. (2016) The role of pluripotency factors to drive stemness in gastrointestinal cancer. Stem Cell Res 16: 349-357.
- Ruiz G, Valencia-Gonzalez HA, Perez-Montiel D, Munoz F, Ocadiz-Delgado R, et al. (2019) Genes Involved in the Transcriptional Regulation of Pluripotency Are Expressed in Malignant Tumors of the Uterine Cervix and Can Induce Tumorigenic Capacity in a Nontumorigenic Cell Line. Stem Cells Int 2019: 7683817.
- Chen Y, Shi L, Zhang L, Li R, Liang J, et al. (2008) The molecular mechanism governing the oncogenic potential of SOX2 in breast cancer. J Biol Chem 283: 17969-17978.
- Yoshida GJ, Saya H (2014) Inversed relationship between CD44 variant and c-Myc due to oxidative stress-induced canonical Wnt activation. Biochem Biophys Res Commun 443: 622-627.
- Yoshida GJ (2018) Emerging roles of Myc in stem cell biology and novel tumor therapies. J Exp Clin Cancer Res 37: 173.
- Welcker M, Clurman BE (2008) FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat Rev Cancer 8: 83-93.
- Dayton TL, Jacks T, Vander Heiden MG (2016) PKM2, cancer metabolism, and the road ahead. EMBO Rep 17: 1721-1730.
- Gupta V, Bamezai RN (2010) Human pyruvate kinase M2: a multifunctional protein. Protein Sci 19: 2031-2044.
- Dong G, Mao Q, Xia W, Xu Y, Wang J, et al. (2016) PKM2 and cancer: The function of PKM2 beyond glycolysis. Oncol Lett 11: 1980-1986.
- Zhang Z, Deng X, Liu Y, Sun L, Chen F (2019) PKM2, function and expression and regulation. Cell Biosci 9: 52.
- Chaneton B, Hillmann P, Zheng L, Martin ACL, Maddocks ODK, et al. (2012) Serine is a natural ligand and allosteric activator of pyruvate kinase M2. Nature 491: 458-462.
- Keller KE, Doctor ZM, Dwyer ZW, Lee YS (2014) SAICAR induces protein kinase activity of PKM2 that is necessary for sustained proliferative signaling of cancer cells. Mol Cell 53: 700-709.
- Zhou Z, Li M, Zhang L, Zhao H, Sahin O, et al. (2018) Oncogenic Kinase-Induced PKM2 Tyrosine 105 Phosphorylation Converts Nononcogenic PKM2 to a Tumor Promoter and Induces Cancer Stem-like Cells. Cancer Res 78: 2248-2261.
- Lv L, Li D, Zhao D, Lin R, Chu Y, et al. (2011) Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell 42: 719-730.
- Anastasiou D, Yu Y, Israelsen WJ, Jiang JK, Boxer MB, et al. (2012) Pyruvate kinase M2 activators promote tetramer formation and suppress tumorigenesis. Nat Chem Biol 8: 839-847.
- Lu W, Cao Y, Zhang Y, Li S, Gao J, et al. (2016) Up-regulation of PKM2 promote malignancy and related to adverse prognostic risk factor in human gallbladder cancer. Sci Rep 6: 26351.
- Tryfonidis K, Papadaki C, Assele S, Lagoudaki E, Menis J, et al. (2018) Association of BRCA1, ERCC1, RAP80, PKM2, RRM1, RRM2, TS, TSP1, and TXR1 mRNA expression levels between primary tumors and infiltrated regional lymph nodes in patients with resectable non-small cell lung cancer. Pharmacogenomics J 19: 15-24.
- Azoitei N, Becher A, Steinestel K, Rouhi A, Diepold K, et al. (2016) PKM2 promotes tumor angiogenesis by regulating HIF-1alpha through NF-kappaB activation. Mol Cancer 15: 3.
- Papadaki C, Manolakou S, Lagoudaki E, Pontikakis S, Ierodiakonou D, et al. (2020) Correlation of PKM2 and CD44 Protein Expression with Poor Prognosis in Platinum-Treated Epithelial Ovarian Cancer: A Retrospective Study. Cancers (Basel) 12: 1013.
- Kuehne A, Emmert H, Soehle J, Winnefeld M, Fischer F, et al. (2015) Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol Cell 59: 359-371.
- James AD, Richardson DA, Oh IW, Sritangos P, Attard T, et al. (2020) Cutting off the fuel supply to calcium pumps in pancreatic cancer cells: role of pyruvate kinase-M2 (PKM2). Br J Cancer 122: 266-278.
- Jin K, Gao W, Lu Y, Lan H, Teng L, et al. (2012) Mechanisms regulating colorectal cancer cell metastasis into liver (Review). Oncol Lett 3: 11-15.
- Kwon OH, Kang TW, Kim JH, Kim M, Noh SM, et al. (2012) Pyruvate kinase M2 promotes the growth of gastric cancer cells via regulation of Bcl-xL expression at transcriptional level. Biochem Biophys Res Commun 423: 38-44.
- Hasan D, Gamen E, Abu Tarboush N, Ismail Y, Pak O, et al. (2018) PKM2 and HIF-1alpha regulation in prostate cancer cell lines. PLoS One 13: 0203745.
- Alsaleh M, Leftley Z, Barbera TA, Koomson LK, Zabron A, et al. (2020) Characterisation of the Serum Metabolic Signature of Cholangiocarcinoma in a United Kingdom Cohort. J Clin Exp Hepatol 10: 17-29.
- Fernandez-Marcos PJ, Nobrega-Pereira S (2016) NADPH: new oxygen for the ROS theory of aging. Oncotarget 7: 50814-50815.
- Guo J, Ren R, Yao X, Ye Y, Sun K, et al. (2020) PKM2 suppresses osteogenesis and facilitates adipogenesis by regulating beta-catenin signaling and mitochondrial fusion and fission. Aging (Albany NY) 12: 3976-3992.
- Wang C, Jiang X, Li X, Song S, Meng Q, et al. (2020) Long noncoding RNA HULC accelerates the growth of human liver cancer stem cells by upregulating CyclinD1 through miR675-PKM2 pathway via autophagy. Stem Cell Res Ther 11: 8.
- Debeb BG, Lacerda L, Larson R, Wolfe AR, Krishnamurthy S, et al. (2016) Histone deacetylase inhibitor-induced cancer stem cells exhibit high pentose phosphate pathway metabolism. Oncotarget 7: 28329-28339.
- Manolakou SD. Ziros ZP, Sykiotis GP (2014) NFE2L2 (nuclear factor, erythroid 2-like 2). Atlas of Genetics and Cytogenetics in Oncology and Haematology, Switzerland (GPS).
- Ziros PG, Manolakou SD, Habeos IG, Lilis I, Chartoumpekis DV, et al. (2013) Nrf2 is commonly activated in papillary thyroid carcinoma, and it controls antioxidant transcriptional responses and viability of cancer cells. J Clin Endocrinol Metab 98: 1422-1427.
- Heiss EH, Schachner D, Zimmermann K, Dirsch VM (2013) Glucose availability is a decisive factor for Nrf2-mediated gene expression. Redox Biol 1: 359-365.
- Mitsuishi Y, Motohashi H, Yamamoto M (2012) The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol 2: 200.
- Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, et al. (2012) Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22: 66-79.
- Zhu J, Bi Z, Yang T, Wang W, Li Z, et al. (2014) Regulation of PKM2 and Nrf2-ARE pathway during benzoquinone induced oxidative stress in yolk sac hematopoietic stem cells. PLoS One 9: 113733.
- Wang X, Zhang F, Wu XR (2017) Inhibition of Pyruvate Kinase M2 Markedly Reduces Chemoresistance of Advanced Bladder Cancer to Cisplatin. Sci Rep 7: 45983.
- Gines A, Bystrup S, Ruiz de Porras V, Guardia C, Musulen E, et al. (2015) PKM2 Subcellular Localization Is Involved in Oxaliplatin Resistance Acquisition in HT29 Human Colorectal Cancer Cell Lines. PLoS One 10: 0123830.
- He J, Xie G, Tong J, Peng Y, Huang H, et al. (2014) Overexpression of microRNA-122 re-sensitizes 5-FU-resistant colon cancer cells to 5-FU through the inhibition of PKM2 in vitro and in vivo. Cell Biochem Biophys 70: 1343-1350.
- Zhao X, Zhu Y, Hu J, Jiang L, Li L, et al. (2018) Shikonin Inhibits Tumor Growth in Mice by Suppressing Pyruvate Kinase M2-mediated Aerobic Glycolysis. Sci Rep 8: 14517.
- Wang Y, Hao F, Nan Y, Qu L, Na W, et al. (2018) PKM2 Inhibitor Shikonin Overcomes the Cisplatin Resistance in Bladder Cancer by Inducing Necroptosis. Int J Biol Sci 14: 1883-1891.
- Babu MS, Mahanta S, Lakhter AJ, Hato T, Paul S, et al. (2018) Lapachol inhibits glycolysis in cancer cells by targeting pyruvate kinase M2. PLoS One 13: 0191419.
- Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, et al. (2013) Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget 4: 584-599.
- Lamson DW, Plaza SM (2003) The anticancer effects of vitamin K. Altern Med Rev 8: 303-318.
- Su Q, Luo S, Tan Q, Deng J, Zhou S, et al. (2019) The role of pyruvate kinase M2 in anticancer therapeutic treatments. Oncol Lett 18: 5663-5672.
- Ning X, Qi H, Li R, Jin Y, McNutt MA, et al. (2018) Synthesis and antitumor activity of novel 2, 3-didithiocarbamate substituted naphthoquinones as inhibitors of pyruvate kinase M2 isoform. J Enzyme Inhib Med Chem 33: 126-129.
- Ning X, Qi H, Li R, Li Y, Jin Y, et al. (2017) Discovery of novel naphthoquinone derivatives as inhibitors of the tumor cell specific M2 isoform of pyruvate kinase. Eur J Med Chem 138: 343-352.
- Ji Q, Ding YH, Sun Y, Zhang Y, Gao HE, et al. (2016) Antineoplastic effects and mechanisms of micheliolide in acute myelogenous leukemia stem cells. Oncotarget 7: 65012-65023.
- An Y, Guo W, Li L, Xu C, Yang D, et al. (2015) Micheliolide derivative DMAMCL inhibits glioma cell growth in vitro and in vivo. PLoS One 10: 0116202.
- Shang D, Wu J, Guo L, Xu Y, Liu L, et al. (2017) Metformin increases sensitivity of osteosarcoma stem cells to cisplatin by inhibiting expression of PKM2. Int J Oncol 50: 1848-1856.
- Silvestri A, Palumbo F, Rasi I, Posca D, Pavlidou T, et al. (2015) Metformin Induces Apoptosis and Downregulates Pyruvate Kinase M2 in Breast Cancer Cells Only When Grown in Nutrient-Poor Conditions. PLoS One 10: 0136250.
- Su Q, Tao T, Tang L, Deng J, Darko KO, et al. (2018) Down-regulation of PKM2 enhances anticancer efficiency of THP on bladder cancer. J Cell Mol Med 22: 2774-2790.
- Zhou Y, Huang Z, Su J, Li J, Zhao S, et al. (2020) Benserazide is a novel inhibitor targeting PKM2 for melanoma treatment. Int J Cancer 147: 139-151.
- De Francesco EM, Sotgia F, Lisanti MP (2018) Cancer Stem Cells (CSCs): Metabolic strategies for their identification and eradication. Biochem J 475: 1611-1634.
- Maeda M, Murakami M, Takegami T, Ota T (2008) Promotion or suppression of experimental metastasis of B16 melanoma cells after oral administration of lapachol. Toxicol Appl Pharmacol 229: 232-238.
- Li J, Li S, Guo J, Li Q, Long J, et al. (2018) Natural Product Micheliolide (MCL) Irreversibly Activates Pyruvate Kinase M2 and Suppresses Leukemia. J Med Chem 61: 4155-4164.
Citation: Manolakou SD, Messaritakis I, Souglakos J (2020) PKM2-Mediated Glycolysis and Redox Equilibrium in Cancer Stem Cells: The Metabostemness Phenomenon. J Stem Cell Res Dev Ther 6: 040.
Copyright: © 2020 Stavroula D Manolakou, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.