Proteomics Applied to Pathogenic Entamoeba histolytica: A Review
*Corresponding Author(s):
Nurulhasanah OthmanInstitute For Research In Molecular Medicine, (INFORMM), Universiti Sains Malaysia, Penang, Malaysia
Tel:+60 04 6534877,
Email:nurulhasanah@usm.my
Abstract
Proteomics approaches have been used in many studies since 1990’s. The state-of-art technologies are growing in this field which spans protein separation, protein digestion and mass spectrometry. In this review, we describe traditional and novel proteomic technologies and their reported applications in the study of Entamoeba histolytica. This parasite causes amoebiasis, a parasitic disease of public health importance in tropical and sub-tropical countries. Amoebiasis is classified under the Neglected Tropical Disease (NTD) by World Health Organization. It affects about 10% of world population. Studies using proteomic tools would be very useful to uncover molecules and markers that address important issues in amoebiasis such as new E. histolytica vaccine and drug target candidates, as well as to further our understanding on the disease pathogenesis.
Keywords
Amoebiasis; Biological markers; Entamoeba histolytica; Proteomic technologies
INTRODUCTION
In the 1990s, the term “proteomics” was created with the combination of the words “protein” and “genomics” [1]. Proteomic studies comprise and quantification of proteins and proteome, as well as protein expression, protein interaction and Post-Translational Modifications (PTMs). Meanwhile, proteome is the complete proteins that is or can be expressed by a cell, tissue, or organism [1]. Therefore, the in-depth exploration of a proteome is arguably more difficult than sequencing [1]. The advancement in proteomics research has led to the development of recent technologies for protein and peptide separations, Mass Spectrometry (MS) analysis and isotope labelling for quantification and bioinformatics. Combined with the rapid growth of protein databases, high-throughput proteomics is becoming a reality [2]. Bottom-up method is one of the commonly used approaches in proteomics, whereby the complex proteins are proteolytically digested into peptides prior to protein analysis and characterization [1]. Figure 1 shows the process of bottom-up proteomics. When the bottom-up method is conducted on a mixture of proteins, it is called shotgun proteomics. The approaches used such as Multidimensional Protein Identification Technology (MudPIT), Isotope-Coded Affinity Tag (ICAT) and isobaric Tag for Relative and Absolute Quantitation (iTRAQ). These methods are potent in generating analytical data [3]. Entamoeba histolytica is a parasitic protozoan that can cause amoebiasis in humans and other animals such as dogs. It is widespread in tropical and subtropical regions of the world with lack of clean water, poor sanitation and hygiene including Mexico, Bangladesh and Vietnam [4-6]. In Malaysia, the prevalence of amoebiasis among disadvantaged communities ranges from 1 to 14% [7]. E. histolytica has two life stages, trophozoite is the active form that induce disease while infectious cyst is the dormant form [6]. The cyst causes excystation once it reaches the small intestine, particularly the ileum, releasing trophozoites, the parasite stage that proliferates in the human large intestine and is responsible for the symptoms of amoebiasis [8]. Researchers mostly conduct studies on E. histolytica infection on the trophozoite stage, which is easy to grow in axenic culture media. Therefore, information on the cyst is very rare due to its inability to duplicate and the lack of reproducibility [8]. Almost 90% of infections are asymptomatic while the remaining 10% exhibit arrange of manifestations such as amoebiasis, dysentery, acute diarrhea, amoebic colitis and Amoebic Liver Abscesses (ALA) [6,9]. Amoebiasis is usually treated with drugs such as metronidazole and tinidazole. However, these drugs can have adverse side effects and are not readily available in certain countries or areas [6]. Moreover, currently there is no vaccine available to prevent amoebiasis. Hence, proteomic technologies can be useful tools in discovery of new biological markers as targets for vaccine and drug developments to treat and prevent amoebiasis. In this review, we highlighted the proteomic strategies and techniques and how they have been used to study E. histolytica proteome. Furthermore, we also identified the advantages and limitations of the technologies as well as provide recommendations for future studies in amoebiasis.
Proteomic Technologies
Protein fractionation: Fractionation or separation of crude protein samples is performed to reduce its complexity and to minimize the interference between peptides in downstream workflow, especially for mass spectrometry analysis. The fractionation or separation methods are based on molecular weight, hydrophobicity and isoelectric point. Fractionation at the protein level according to the molecular weight and isoelectric point using SDS-PAGE (Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis) and Isoelectric Focusing (IEF) are commonly used in proteomics. In a study by Mostovenko et al., SDS-PAGE method showed better coverage of the proteome compared to IEF, while it also provided the molecular weight information of the proteins [10].
Multiple fractionations such as Two-Dimensional Electrophoresis (2-DE or 2D-PAGE) are commonly used for complex proteins mixtures since they facilitate proteomics analysis and improve the proteome coverage [10]. It is performed by coupling two methods involving IEF, followed by SDS-PAGE. This allows the separation of complex mixtures of proteins according to their isoelectric point, molecular weight, solubility and relative abundance [11]. It can categorize more than 5000 proteins simultaneously and detect less than 1mg protein per spot based on the gel size and pH gradient [11]. After the separation, the protein spots are excised, digested and then subjected to MS analysis for protein identification.
Although 2D-PAGE is a powerful method which allows concurrent visualization of large portions of proteomes, it has limited use as a quantitative tool due to the low sensitivity and labelling capability of the reagents [12]. Moreover, only a few recent efforts have been directed at comparing different species or strains of organisms [13]. When involving cross species, it is difficult for post-analysis using 2D-PAGE. The protein spot location precisely indicates the coordinate of both species due to differences in isoelectric point and molecular weight. However, it can be affected by the genomic differences resulting in amino acid substitutions, splice variants, post-translational modifications, truncations or insertions [13]. This led to the discovery of Two-Dimensional Fluorescence Difference Gel Electrophoresis (2D-DIGE) adding high accuracy of quantitative dimension which allows multiple protein extracts to be separated on the same gel [14]. This is eased by labelling the extracts using CyDye DIGE fluorescent which is resolvable, and the dyes are size and charge-matched [12]. Hence, the 2D-DIGE would be the best choice for gel-based approach as it overcomes the drawbacks of the traditional 2D-PAGE.
Furthermore, the classic 2D-PAGE might raise unavoidable challenges such as decreased enzyme accessibility to the protein, low protein coverage due to ineffective large peptides extraction from the gel, and the need to identify hundreds of distinct spots. These complications have been addressed with the advancement of gel-free technologies [15]. They have been shown to be effective tools in current proteomic studies. Equipment such as Agilent 3100 OFFGEL Fractionator is commonly used to separate complex samples into discrete liquid fractions on Immobilized pH Gradient (IPG) strips, corresponding to their Isoelectric Point (pI) by IEF. Another example is Gelfree 8100 Fractionation Station (Expedeon Ltd, Swavesey), where it is capable to fractionize complex protein samples based on their molecular weight [16]. One of the common advantages of both fractionation systems is the convenience of liquid phase protein/peptide recovery which reinforces the downstream workflow flexibility [17].
Protein digestion: In a typical experiment, proteins are digested prior to MS analysis of the resulting peptides [18]. To produce a peptides mixture comprising a high percentage of amino acid sequences, an effective protein digestion is needed and one of the most commonly used methods is in-gel digestion [18]. This method was established by Rosenfeld et al., comprised of destaining, reduction, and alkylation of cysteines, enzymatic cleavage of proteins into peptides and extraction of peptides from the gel [18]. For ESI-MS, the procedure includes an additional desalting step which is optional for MALDI-MS [19]. The buffers used to extract proteins often contain salts that form with proteins, which could complicate data interpretation and suppress ionization during ESI-MS analysis. Therefore, proteins need to be separated from non-volatile salt prior to mass spectrometry analysis [20]. A distinct number of representative peptides with various molecular masses are acquired for the in-gel digestion of proteins with specific proteases such as trypsin, chymotrypsin, Lys C and Glu C [21]. The masses of these peptides are then used to identify the proteins by mass-spectrometry. Large proteolytic peptides are not easy to recover and analyzed by in-gel digestion [22]. Thus, in-solution digestion is an alternative way to recover large proteolytic peptides. Besides, it gives better sequence coverage and improved probability to detect functionally significant protein modifications [22]. Furthermore, in-solution digestion has an extensive selection of proteinases compared to in-gel approaches. Thus, it increases the flexibility of proteolytic specificity. Figure 2 illustrates the in-gel digestion and in-solution digestion.
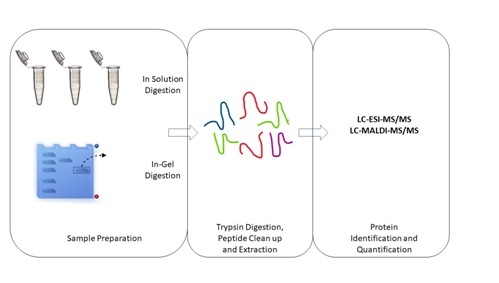
Figure 2: In-gel and in-solution digestions.
Peptide fractionation using liquid chromatography coupled with tandem Mass Spectrometry (MS/MS): The combination of several fractionation steps is necessary to overcome sample complexity and its wide dynamic characteristics [1]. This can be performed by pre-fractionating protein either on-gel or off-gel, followed by Liquid Chromatography (LC) to separate the peptides. Liquid Chromatography Tandem Mass Spectrometry (LC-MS/MS) has made feasible and effective methodology for shotgun proteomics by pre-fractionating the peptides mixture prior to data collection using MS/MS, thus addressing the problem of sample complexity [23].
Reverse-Phase (RP) Liquid Chromatography (LC) is commonly employed to resolve peptides mixture based on their hydrophobicity under gradient condition by changing the gradient slope, or the organic solvent composition of the mobile phase [24]. This can improve protein identification by mass spectrometry.
Mass spectrometry is a powerful tool for proteomics research as it allows more complete characterization of protein isoforms and Post-Translational Modifications (PTM) [25]. Mass spectrometry basically ionizes the analyte molecules into gas-phase ions, which is then separated based on the mass-to-charge-ratio (m/z) by a mass analyzer. Finally, the number of ions is recorded by a detector at each m/z value [25]. Currently, the rapid growth of new methods and advancement in mass spectrometry technologies have improved the resolution, mass accuracy, sensitivity and scan rate of the instrument for enhancing the protein identification and quantification [1].
Peptide ionization is the conversion of peptides into gas-phase ions. The common ionization methods in mass spectrometry for shotgun proteomics are Electrospray Ionization (ESI) and Matrix-Assisted Laser Desorption/Ionization (MALDI) [26]. Both ionization methods are usually coupled with pre-fractionation methods such as RP-LC separations or gel-based separations. MALDI is generally interfaced with the Time-Of-Flight (TOF) mass analyzer, which has a wide range of mass-to-charge ratio and is compatible with large singly charged ions [26]. This method is relatively robust, sensitive and the presentation of the data is simple to interpret due to the generation of mainly singly charged ions [27]. MALDI involves desorption of solid-phase analytes to ionize the sample. However, this action requires significant energy input, therefore a matrix is used to protect the sample from direct effects from the energy, resulting in a moderate desorption of analyte molecules from the surface, forming singly charged ions [26]. Unlike MALDI, ESI does not require matrix because the liquid samples are directly introduced into the mass analyzer in the nebulized form, resulting in charged droplets which undergo desolvation, thus forming gas-phase and multiply charged analytes [26].
Identification of peptide is obtained by comparing the tandem mass spectra derived from peptide fragmentation with theoretical tandem mass spectra generated from in silico digestion of a protein database [28]. Peptide fragmentation mechanism such as Collision-Induced Dissociation (CID) is one of the most commonly used in tandem MS [29]. Here, individual peptide ions are isolated, fragmented and the masses recorded to obtain partial or complete information. This process is commonly known as tandem MS or MS/MS. This method demands the acceleration of kinetic energy of ions for collision with residual gas that triggers the cleavage of peptide bonds (or C-N bonds) at the lowest energy pathways. Thus, a series of b-fragment ions and y-fragment ions is obtained from the peptides fragmentation [1,28,30]. From the tandem MS spectrum of a particular peptide generated, partial primary sequence can be determined by the comparison of major peaks in the spectrum generated with theoretical molecular masses of the amino acid monomers within the peptide [31]. Protein identification from a complex mixture is accomplished by matching the mass of the parent ion derived from peptide fragmentation against tandem mass spectra and all candidate peptides of in silico protein sequence databases [1]. The database-searching algorithms such as MASCOT, Paragon™ Algorithm, or theoretical spectra calculated from known DNA or protein sequence database are based on the m/z of the precursor ions [30,32-34]. Some examples of the well-known database search engines that are frequently used are UniProt/Swiss-Prot and NCBI sequence database [35,36].
Proteomics strategies and techniques applied to E. histolytica: The 2D-PAGE approach has been utilized in many proteomic studies on E. histolytica [13,37-40]. Using this technique, Leitsch et al., compared the proteome profiles of E. histolytica and the related but non-pathogenic E. dispar [38]. The result showed that 2D- PAGE was able to separate thousands of proteins on a single gel [38]. The authors then identified 1547 E. histolytica and 1583 E. dispar protein spots. Their results also showed 1553, 1657, 1139 and 571 protein spots of E. histolytica for pH 3-10, 5-8, 4-7 and 7-10, respectively [38]. However, the authors noted that 2D-PAGE was time consuming and labor- intensive, with significant gel to gel variation. Similarly, in 2009, Davis et al., compared the proteomes of E. histolytica HM1:IMSS and E. dispar SAW760 using 2D-PAGE and in each biological replicate of the species, authors could resolve an average of 2676 protein spots[13].
Protein separations by OFFGEL fractionator have been utilized in several proteomic studies on E. histolytica, such as analysis of excretory-secretory proteins of E. histolytica by Wong et al., characterization of E. histolytica antigenic proteins by Huat et al., investigation of E. histolytica antigenic proteins in Amoebic Liver Abscess (ALA) aspirates by Othman et al., and analysis of E. histolytica crude antigen for serodiagnosis of ALA by Ning et al., [41-44].
OFFGEL fractionation is also applied in a previous study where the Crude Soluble Antigenic (CSA) proteins were extracted from E. histolytica trophozoites and separated by Agilent 3100 OFFGEL Fractionator, on IPG strip of pH 3 to 10 in a 12-wells setup, followed by second dimension fractionation by SDS-PAGE [42]. The targeted 75 kDa antigenic protein was excised from SDS-PAGE gel for in-gel tryptic digestion and subjected to Electrospray Ionization Mass Spectrometry (ESI-TRAP) via the Ultimate 3000 nano HPLC system (Dionex) coupled with 4000 QTRAP mass spectrometry (Applied Biosystems) for protein identification. The targeted protein was identified as E. histolytica acetyl-CoA synthetase with protein and peptide scores of 294 and 1456, respectively [42]. A similar approach was performed by Ning et al., for the evaluation of CSA proteins from E. histolytica [44]. The mass spectrometry analysis identified the 70 k Da antigenic protein as Phosphoglucomutase (PGM) with the protein and peptide scores of 793 and 19, respectively. Furthermore, the identified PGM was shown to be potentially useful for ALA diagnosis in the study. Overall, the above studies show that the combination OFFGEL and SDS-PAGE offers a better protein separation and enable confirmation of isolated target proteins [44].
Recent proteomic studies on E. histolytica have been performed using mass spectrometry, for example, MALDI-TOF/TOF and LC-MS/MS [13,37,40,41,45]. In 2007, Tolstrup and colleagues demonstrated the usage of MALDI-TOF mass spectrometry for in-gel based proteomics analysis of E. histolytica trophozoites (HM1:IMSS), and they successfully identified 63 proteins that were predicted to be associated with the cytoskeleton, surface, glycolysis, RNA/DNA metabolism, the ubiquitin-proteasome system, vesicular trafficking and signal transduction [40].
A differential protein expression study between E. histolytica and E. dispar was conducted by Davis et al., to identify the differentially expressed proteins between the two species. In their study, the whole cell lysates of two species were separated using 2-DE [13]. The targeted protein spots were then in-gel digested into peptides, followed by protein identification via MALDI-TOF/TOF (ABI 4700). They found three proteins which showed higher protein expression in E. histolytica HM1: IMSS and one showed higher protein expression in E. dispar, with a minimum of 5-fold or greater cut-off [13]. One of the differentially expressed proteins was E. histolytica Alcohol Dehydrogenase 3 (EhADH3) which may play an important role in the disease pathogenesis [13].
Meanwhile, a proteomics analysis on the dynamic changes of E. histolytica phagosome proteins was performed by Okada et al., [46]. In their study, the phagosome peptides were separated and identified by LC-MS/MS system that consisted of Finnigan LCQ ion trap mass spectrometer with Protana nanospray ion source interfaced to a self-packed Phenomenex Jupiter C18 reversed-phase capillary column. They successfully detected 159 proteins from the analysis, which comprised of 103 proteins with known or predicted functions, while the remaining 56 were hypothetical proteins [46]. The proteomic data on kinetics and strain variation of phagosome biogenesis from this study gave a basis of knowledge on phagosome biogenesis [46]. This may lead to further understanding of the functions of phagocytosis in this parasite. Furthermore, it also can help to facilitate functional assignment of individual hypothetical proteins localized to phagosomes, which is essential for annotation of the genome database [46].
In a ESA profiling study conducted by Ujang et al., a total of 219 excretory-secretory proteins were identified, in which 18 and 112 proteins were uniquely identified by LC-MALDI- TOF/TOF and LC-ESI-MS/MS, respectively [45]. Meanwhile, 89 proteins were identified by both systems. The result shows the LC-ESI-MS/MS was more sensitive when compared to LC-MALDI-TOF/TOF for protein identification in this study [45]. In their study, 27% of the proteins were involved in metabolic processes, 35% of the proteins were found to be involved in catalytic activity, and 21% were associated with the cell parts [45]. The utilization of advance proteomics technology in this study has led to a better identification of E. histolytica excretory-secretory proteins.
Proteomics technology was also applied in a cell surface proteome study of E. histolytica using biotinylated proteins by Biller et al., [47]. The gel containing separated protein bands were excised and in gel digested into peptides prior to LC-MS/MS. The digested peptides were subjected to RP-LC for the separation, coupled with LTQ-Orbitrap XL mass spectrometer for protein identification. A total of 693 putative surface-associated proteins were identified. Among them, in silico analysis predicted about 26% were membrane-associated proteins, as they contained transmembrane domains and/or signal peptides. Additional of 25% represent nonclassical secreted proteins. There was no membrane association sites predicted for the remaining 49% of the identified proteins. For verification, 23 proteins were randomly selected and analyzed by immunofluorescence microscopy. Definite surface localization was showed by 20 (87%) proteins. These findings specify that a far greater number of E. histolytica proteins than earlier supposed are surface-associated, a phenomenon that may be based on the high membrane turnover of E. histolytica.
Furthermore, proteomics analysis was also performed to illustrate and characterize the endomembrane of E. histolytica [48]. The cell fractionation and an extensive proteomic analysis to search for principal components of the endomembrane system in E. histolytica were the aim of this study. The internal membrane protein fractions were tryptic digested and separated by reverse-phase liquid chromatography HPLC, coupled with LTQ Orbitrap Velos for the mass spectrometry analysis. The LC-MS/MS analyses identified a total of 5683 peptides matching with 1531 proteins, which corresponded to approximately 20% of the total amoebic proteome [48]. Over 1500 proteins were identified, and the two top categories contained components of trafficking machinery and GTPases. There are over 100 markers from the Endoplasmic Reticulum (ER), Golgi, multivesicular bodies MBV and retromers. This data represented the in-depth proteomics analysis of subcellular compartments in E. histolytica and allowed a detailed map of vesicle traffic components in an ancient single-cell organism that lacks a stereotypical ER and Golgi apparatus to be established [48].
In a previous report, Luna-Nácar et al., used LC-MS/MS to study the proteome of E. histolytica trophozoites, cysts and in vitro-produced cyst-like structure CLS in order to determine the nature of CLS which contributed the new knowledge on the cyst stage, and identify possible proteins and pathways involved in the encystment process [8]. In their findings, about 70% of CLS proteins were shared with trophozoites, even though differences were observed in the relative protein abundance [8]. While trophozoites showed a greater abundance of proteins associated to a metabolically active cell, CLS showed higher expression of proteins related to proteolysis, redox homeostasis, and stress response [8]. They also suggested that encystment and CLS formation could be distinct stress responses [8].
To fulfil the gap of knowledge on oxidized proteins in E. histolytica, Shahi et al., performed a large-scale identification and quantification of the oxidized proteins in oxidatively stressed E. histolytica trophozoites using resin-assisted capture coupled to LC-MS/MS [9]. They have detected 154 Oxidized Proteins (OXs) and the functions of some of these proteins were associated with antioxidant activity, maintaining the parasite's cytoskeleton, translation, catalysis, and transport [9]. They also found that oxidation of the Gal/GalNAc impairs its function and contributes to the inhibition of E. histolytica adherence to host cells and provide evidence that arginase, is involved in the protection of the parasite against oxidative stress [9].
Following the discovery of potential biological marker from E. histolytica, validation should be performed to explore its usefulness especially for those that will be used in clinical studies. This can be facilitated by the use of mass targeted approach or Multiple Reactions Monitoring (MRM).
FUTURE PROSPECTIVE
Previous proteomic studies have shown the usefulness of these technologies to elucidate the proteome of E. histolytica [13,37,40,41,45]. Besides, some of the reported proteins molecules such as the differentially expressed proteins and/or membrane proteins of E. histolytica could be potentially utilized in future study as targets for new vaccine and drug developments as these proteins play important roles in the disease pathogenicity based on their functionality [6]. Other than the gel-based proteomics, we would recommend the non-gel based quantitative proteomics applications using chemical labelling such as iTRAQ (Isobaric Tag for Relative and Absolute Quantitation) for E. histolytica study. iTRAQ gains its popularity nowadays in many differential protein expression studies due to its high accuracy, relevance, precision and the impaired quantification efficiency that generates reproducible and high-throughput results for biological interpretation. The chemical labelling would be the great approach on differential protein expression studies in the foreseeable future.
In fact, it would be interesting to investigate further the uncharacterized or also known as hypothetical proteins from previous studies. This can be accomplished by bioinformatics analysis to elucidate the virtual structure of the molecules and further laboratory analysis by the crystallography. These proteins perhaps hold promising potential as new targets for prevention or treatment of amoebiasis.
CONCLUSION
The advancements in the proteomics technologies are very useful for scientists in the biomedical field to discover new biological markers which could be utilized for diagnosis, treatment and prevention of amoebiasis. Furthermore, the findings also help us to better understand the disease pathogenesis at the molecular level.
ACKNOWLEDGEMENT
We would like to express our gratitude to the research funding from Universiti Sains Malaysia RUI grant, No. 1001/CIPPM/812118. Ng Yee Ling received financial assistance from USM Graduate Assistant Scheme.
REFERENCES
- Biller L, Matthiesen J, Kühne V, Lotter H, Handal G, et al. (2014) The cell surface proteome of Entamoeba histolytica. Mol Cell Proteomics 13: 132-144.
- Perdomo D, Aït-Ammar N, Syan S, Sachse M, Jhingan GD, et al, (2015) Cellular and proteomics analysis of the endomembrane system from the unicellular Entamoeba histolytica. J Proteomics 112: 125-140.
Citation: Kumarasamy G, Ng YL, Noordin R, Othman N (2018) Proteomics Applied to Pathogenic Entamoeba histolytica: A Review. J Protein Res Bioinform 1: 001.
Copyright: © 2018 Gaayathri Kumarasamy, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.