The Four Stages of Insulin Resistance Induced Chronic Diseases of Aging
*Corresponding Author(s):
Deepak ChopraThe Chopra Foundation, Orlando, United States
Tel:+1 9174149188,
Email:frank@deepakchopra.com
Abstract
There is a fundamental connection between mitochondrial function, bioenergetics and insulin signaling. The dysfunction of mitochondria plays a causative role in metabolic and chronic diseases due to bioenergetic capacity impairment. In turn, insulin resistance is a major pathophysiological basis for type 2 diabetes, other metabolic syndromes and cardiovascular disease. Insulin resistance and hyperinsulinemia is increasingly recognized as being correlated with accelerated cognitive decline and Alzheimer’s disease, as well as cancers of reproductive and gastrointestinal tissues. The connection between insulin resistance and mitochondrial dysfunction indicates their fundamental involvement in chronic diseases of aging. In this paper we provide an up-to-date review of insulin resistance and its connections to a number of pathologies as well as phases of insulin resistance progression.
Introduction
Whole-body energy homeostasis requires a balance between food intake and energy expenditure. Glucose and fatty acids are major sources of energy. Cellular and systemic glucose metabolism is predominantly maintained by insulin and glucagon. After feeding, glucose enters the bloodstream through the gut and arrives at the pancreas where it stimulates insulin secretion from pancreatic beta cells. Insulin is an endocrine hormone that acts on peripheral tissues such as skeletal muscle, adipose tissue, and the liver. Therefore, insulin increases glucose uptake in adipocytes and myocytes and suppresses hepatic glucose production (via gluconeogenesis) while promoting hepatic glycogenesis and lipogenesis. Moreover, insulin inhibits lipolysis (lipid metabolism) in white adipose tissue (WAT), which results in a decrease in free fatty release from WAT. Conversely, during fasting, when blood glucose levels are low, glucagon is released from pancreatic alpha cells to maintain normal blood glucose levels. Glucagon is an endocrine hormone, which elevates blood glucose levels by promoting hepatic glucose production using gluconeogenesis and glycogenolysis. Decreased circulating insulin and increased glucagon levels during fasting promote lipolysis in WAT and release free fatty acids into the bloodstream to provide energy substrate to such tissues as the heart and skeletal muscles. Thus, insulin and glucagon act mutually antagonistically to maintain whole-body energy homeostasis at cellular and systemic levels during feeding and fasting conditions.
Most eukaryotic cells use glucose as a primary source of energy. A certain amount of glucose is needed for cells' functioning. However, elevated glucose levels can be toxic. Glucose metabolism is tightly regulated and coordinated at cellular and systemic levels. At the physiological level, glucose metabolism is regulated by the endocrine action of polypeptide hormones, such as insulin and glucagon. At the cellular level, it is regulated by energy sensors, involving AMP-Activated Protein Kinase (AMPK), sirtuin, and mTOR pathways. Defects in any of these pathways can lead to the deregulation of glucose metabolism and eventually result in the development of diabetes. Cellular bioenergetics plays a crucial role in maintaining systemic glucose homeostasis. Excess glucose present in the circulation can lead to harmful effects. Elevated blood glucose levels after the meal cause pancreatic beta cells to increase glucose uptake and metabolize it to generate ATP through oxidative phosphorylation. The resulting elevated ATP/ADP ratio works as an important signal responsible for secretion of insulin by beta cells [1,2]. Thus, insulin instructs metabolic organs to respond to elevated blood glucose levels and restore the optimum concentration, which is critical since both hypoglycemia and hyperglycemia can cause major complications. Insulin not only promotes glucose uptake in target tissues, but also regulates mitochondrial function. Insulin increases mitochondrial oxidative capacity, mitochondrial biogenesis, and coupling efficiency of oxidative phosphorylation [3,4]. Insulin also regulates the molecular clocks to the circadian rhythm of the fasting/feeding cycle [5].
Chronic overnutrition leads to mitochondrial dysfunction and Insulin Resistance (IR). Nutrient mismanagement causes the accumulation of lipid intermediates such as Diacylglycerol (DAG) and/or ceramides causing inflammation and Reactive Oxygen Species (ROS) in muscle. These intermediates are harmful to cellular function and can cause mitochondrial dysfunction. Therefore, mitochondria play a crucial role in maintaining a well-balanced nutrient metabolism. Impaired glucose utilization manifests, for example, as lower insulin sensitivity in muscle [6].
Energy availability is fundamental to survival. When energy sources become unavailable, IR can become an adaptation that promotes survival. It can prevent glucose utilization by peripheral tissues to make sufficient amounts of glucose available for brain metabolism. IR also occurs during normal physiological processes, such as puberty, pregnancy, and physical/physiological stress, where it may help the body conserve energy. Following a periodic cycle, such as the feeding/fasting cycle, IR is a healthy event playing a critical role in healthy physiology and metabolic flexibility. However, pathological IR contributes to the onset of chronic diseases of aging such as Alzheimer’s Disease (AD), accelerated cognitive decline, heart and vascular disease, and various types of cancer.
Insulin is involved in approximately 1700 signaling pathways, many of which are intertwined. Insulin signaling pathways promote cell survival, growth and replication, when nutrient availability is balanced with the capacity of bioenergetic machinery, i.e. healthy mitochondria. When an imbalance arises, numerous stress pathways are activated. Other mechanisms, such altered composition of gut microbiota and disruptions to the synchronization of circadian cycles may contribute to IR and can be major mediators of metabolic stress responses.Five major related defects in insulin signaling have been identified as: (1) overactive adipose tissue lipolysis, (2) pancreatic alpha cell hypersecretion of glucagon, (3) renal tubule hyper reabsorption of glucose, (4) impaired postprandial secretion of intestinal incretins, and (5) insulin resistance in the brain [7].
Insulin has important roles in lipid metabolism suppressing adipose lipolysis, stimulating hepatic lipid synthesis, and inhibiting liver conversion of lipids into ketones. In insulin resistant states, the ability of insulin to promote energy storage is impaired. When peripheral tissues become insulin resistant, impaired glucose uptake contributes to postprandial hyperglycemia (e.g., glucose intolerance).The development of IR in the liver often leads to uncontrolled hepatic glucose production. Increased flux of fatty acids and glycerol from insulin resistant adipose tissue accompanying high circulating insulin levels drive the paradox of hepatic insulin resistance with increased lipid synthesis in the liver.
Adipose tissue is the central endocrine “organ” regulating metabolic homeostasis and hence overall health. As obesity develops, healthy expansion of adipose tissue can preserve insulin sensitivity and metabolic health. However, when adipose tissue becomes hypoxic, inflamed, and unable to efficiently store the excess lipid, IR and metabolic syndrome develops. Lipids then accumulate in non adipose tissues, forming ectopic fat deposits mainly in the liver, skeletal and cardiac muscle, pancreas, and the brain. They become a major cause of inflammation, mitochondrial dysfunction, and lost synchronization of circadian insulin signaling within and between organs and tissues of the body. Obesity is a major aspect of the insulin-insensitive adult form of diabetes with central abdominal fat being an important risk factor for type 2 diabetes. Fat cell hypertrophy is responsible for impaired insulin sensitivity and carbohydrate tolerance [8].
Regional distribution of central abdominal fat is connected to adverse effects of lipid and glucose metabolism, including a risk of type 2 diabetes [9]. Subcutaneous adipocyte size in the abdomen appears to be the best predictor of type 2 diabetes [10]. Overgrown adipocytes with impaired perfusion become dysfunctional, leading to hypoxia and inflammation [11-13]. These changes can be detected before the onset of IR [14] indicating that measurements of hypoxia and inflammation can be useful in predicting IR and hence preventing type 2 diabetes. Local insulin signaling is disrupted within the adipocyte, leading to hyperlipidemia and ectopic seeding of fat in organs and tissues that are not equipped to serve as lipid storing depots [15]. Abolishing localized insulin signaling in adipocytes leads to transient IR, fatty liver, high blood sugar, and high blood insulin. IR is a major pathophysiological basis for type 2 diabetes and other metabolic syndromes, and Cardiovascular Disease (CVD) [7,16,17].
The primary defect in insulin resistant tissue is that insulin is unable to promote glucose transport from the bloodstream into insulin sensitive tissues [18-20]. This is supported by evidence that IR is linked with decreased mitochondrial size, reduced activity of mitochondrial enzymes, and decreased fatty acid oxidation [21-23] and lowered rates of mitochondrial ATP synthesis [24-26]. Consistently, type 2 diabetic patients exhibit low mitochondrial respiration [27,28].
A direct correlation between IR and hypertension has been known for three decades [29]. Hypertension due to IR is linked to renal salt retention and atherogenic dyslipidemia and often leads to cardiovascular disease. IR and hyperinsulinemia has been linked to procoagulant and proinflammatory states, high blood levels of uric acid, ovarian hyperandrogenism, central obesity, Polycystic Ovarian Syndrome (PCOS), obstructive sleep apnea, hepatic nonalcoholic steatosis, many gastrointestinal and reproductive tumors, Alzheimer’s disease, and non-Alzheimer’s disease dementias (Figure 1).
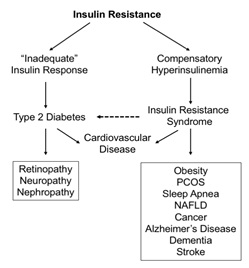 Figure 1: Insulin resistance is implicated in various pathologies ranging from cardiovascular disease to cancer to organ damage to neurological changes [30]. PCOS = polycystic ovarian syndrome; NAFLD = non-alcoholic fatty liver disease.
Figure 1: Insulin resistance is implicated in various pathologies ranging from cardiovascular disease to cancer to organ damage to neurological changes [30]. PCOS = polycystic ovarian syndrome; NAFLD = non-alcoholic fatty liver disease.
The pathogenesis of total body IR in specific tissues may be a heterogeneous multi-factorial process. IR typically occurs in the pancreas as well as in areas in the brain where it can result in impaired satiety, contributing to obesity. Also, metabolic dysfunction in the hippocampus is linked to accelerated cognitive decline and AD when coupled with IR and type 2 diabetes.
Genetics can predispose individuals to pathological IR, for example lean, active adolescents with pathologically insulin resistant parents have mitochondria that are subtly dysfunctional [31], which can further cause skeletal muscle IR, hyperinsulinemia, and dysregulation of insulin receptor function. Risk factors for IR and hyperinsulinemia include genetic susceptibility, visceral obesity, mitochondrial dysfunction, food additives, high dietary saturated fat, high glycemic load, high caloric intake and subclinical endotoxemia. IR is also a risk factor for oncogenesis mediated by inflammation coupled with the formation of reactive oxygen species. Consequently, oxidative modification of DNA can induce mutations, which may be carcinogenic.
First Phase: Skeletal Muscle Insulin Resistance
Skeletal muscle is the primary tissue for IR. Skeletal muscle insulin resistance is due to a defect in glycogen synthesis consequential to impaired Insulin-Stimulated Glucose Uptake (ISGU). The mitochondrial stage of glucose oxidation is more efficient than fatty acid oxidation, which burns cleaner than fatty acids, defined by a lower ratio of oxygen consumption to ATP produced. When glucose cannot be oxidized in the mitochondria, there is overreliance on fatty acids, thus generating greater ROS-mediated redox stress and a core of self-amplifying loops characteristic of metabolic inflexibility.
We call attention tofive core binary self-amplifying instability loops, which promote the “wear and tear”, or “physiological cost” of allostasis. Allostasis or stability through change, in terms of physiology can be defined as stability of vital organ system functions that maintain homeostasis, mediated by the stress response, hormonal and sympathetic nervous system branches, and immune system parameters. Accordingly, these are responsible for reducing stress resilience. The loops are listed below:
- Redox stress/mitochondrial dysfunction
- Mitochondrial dysfunction/insulin resistance
- Insulin resistance and hyperinsulinemia (between tissues)
- Redox stress/inflammation
- Inflammation/psychogenic stress
The final loop underscores how the mind-body connection links to the interlocking cascades of the core instability loops in disease pathogenesis.
Perceived (mental) stress induces the intrinsic allostaticstress response mediated by the hormonal and autonomic branches arising from the midbrain and brainstem. Prolonged mental stress in turn induces physical and circadian stress behaviors (poor diet, sleep and activity).When prolonged; both the extrinsic stress behaviors and the intrinsic allostatic physiology promote proinflammatory cytokines that reduce the threshold for the perception of stress, thus forming a feedforward self-amplifying loop, which may have its origins in either perceived stress or alternatively physical proinflammatory mediators. This is a multi-system pathological IR process with four main phases we discuss here, which can be temporally overlapping.
Young, lean, insulin-resistant individuals demonstrate a decreased capacity for mitochondrial oxidation [31,32] with similar changes observed in aged patients with insulin resistance [33]. This can lead to glucose tolerance driving hepatic lipogenesis and promoting hepatic steatosis [34] but can be significantly improved with exercise. Patients with congenital lipodystrophies lack fat tissue, but show marked lipid accumulation in the liver, which can be reversed with leptin therapy [35]. The resultant negative calorie balance leads to a reduction in ectopic lipid accumulation and improvements in insulin sensitivity [31]. Resistive exercise training, particularly in conjunction with novel approaches for enhancing carnitine availability to skeletal myocytes, appears promising for improving muscle insulin resistance [36].
Importantly, it has been proposed that ceramides could be the ‘new cholesterol’ playing the role of a biomarker in the prediction of heart disease and diabetes risk [37]. Total and Low-Density Lipoprotein (LDL) cholesterol, insulin resistance, visceral adiposity, type 2 diabetes, and CVD are well correlated with blood ceramide levels [38-41]. The proinflammatory action of ceramides promotes ROS and the formation of oxidized LDL within atherosclerotic lesions. When ROS exceed a threshold such as with high levels of ceramides in vascular endothelial and smooth musclecells, the physiological balance of vascular tone mediated by such regulators as nitric oxide, prostacyclin, thromboxane A2, endothelial 1 and angiotensin 1, becomes interrupted and endothelial dysfunction ensues [42-45].
The inflammatory nature of ceramides, characteristic of IR, causes activation of endothelial cell-bound Toll-Like Receptor 4 (TLR-4) promoting inflammatory cytokines. Moreover, ceramides exacerbate the risk of major cardiac events such as fatal and non-fatal heart attacks, sudden death, and stroke by promoting platelet activation and endothelial dysfunction due to the impairment of nitric oxide-mediated vasodilation. Ceramide lowering strategies have been shown to improve IR and hyperinsulinemia manifestations of metabolic syndromes and atherogenesis [46-51]. However, low ceramide levels cause a loss of metabolic homeostasis in tissues, which signifies a hormesis effect, that the mid-range is optimal while too little or too much may generate pernicious effects. Similar to adiposity, too little (lipodystrophy) or too much is detrimental to metabolic health.
Adiponectin is one hormone that helps to regulate ceramide levels and energy homeostasis. It’s beneficial effects are largely due to its positive influence on the degradation of ceramide [52]. In the liver, adiponectin inhibits ceramide-mediated antagonism of insulin signaling, and hyperglycemia. In skeletal muscle it improves ceramide-induced impairment of glucose uptake and thus glucose intolerance. Excessive ceramide can promote mitochondrial dysfunction-mediated apoptosis in other tissues such as the heart, pancreas and brain to pathogenetically underpin cardiomyopathy, insulinopenia, diabetes, Alzheimer’s disease, and Parkinson’s disease.In the liver, skeletal muscle and ectopic sites of lipid deposits, adiponectin enhances fatty acid oxidation. Its inherently insulin sensitizing function highlights the dichotomy of metabolically healthy versus unhealthy obese phenotypes [53].
The decreased capacity to oxidize fuels causes the accumulation of lipid mediators such as ceramides and DAG that leads to IR. A crucial mechanism that impairs substrate oxidation is based on the fact that too many electrons flood into the electron transport system in the mitochondria causing electron leakage towards oxygen and ROS formation. ROS oxidatively modify the molecular chemistry of mitochondria, further impairing bioenergetics with feedforward pathogenic accumulation of reactive ceramide and DAG lipid species, inducing insulin resistance.
A critical feature in insulin resistance and type 2 diabetes is metabolic inflexibility.Healthy individuals can flexibly utilize fatty acids for energy during fasting or low glucose availability, and can switch to using glucose when available. However, type 2 diabetics do not have this degree of metabolic flexibility; they cannot easily switch from fatty acid oxidation to glucose oxidation [54].
Classical views on metabolic flexibility are based on the ability of skeletal muscle to nimbly switch to carbohydrate from lipid oxidation as the fuel source for ATP production at the transition of the fasting to the feeding phase. According to a modern viewpoint, adaptable fuel sourcing in all metabolically flexible tissues is mediated by insulin and FOXO signaling (Figure 2).
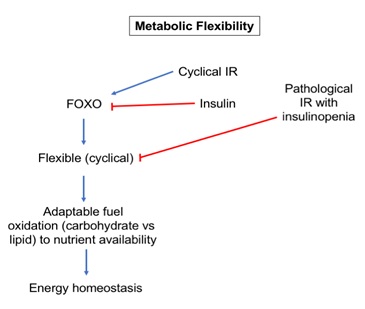 Figure 2: Under healthy conditions, cyclical insulin resistance is adaptive and promotes FOXO-mediated stress resistance programs with energy and redox homeostasis. Under pathologic conditions of insulin resistance, a lack of insulin causes FOXO to be constitutively activated leading to an increase in hepatic glucose output, decreased glucose uptake in skeletal muscle, and decreased insulin secretion from the pancreas.
Figure 2: Under healthy conditions, cyclical insulin resistance is adaptive and promotes FOXO-mediated stress resistance programs with energy and redox homeostasis. Under pathologic conditions of insulin resistance, a lack of insulin causes FOXO to be constitutively activated leading to an increase in hepatic glucose output, decreased glucose uptake in skeletal muscle, and decreased insulin secretion from the pancreas.
In metabolically flexible individuals, meals can cause wide swings in the respiratory quotient (RQ) with relatively minimal changes in insulin secretion required to maintain normal blood glucose concentrations (euglycemia). With pathological transition to non-cyclical insulin resistance, these rhythms become desynchronized and metabolism becomes inflexible. A characteristic sign of pathological IR and type 2 diabetes is the loss of adaptive metabolic flexibility, such that mitochondrial enzymes cannot appropriately adjust to changes in nutrient source. RQ can be used to assess impairment of metabolic flexibility in insulin-resistant muscle tissue [55]. As IR worsens, crosstalk between tissues becomes impaired.
Second Phase: Systemic Insulin Resistance And Hyperinsulinemia
The second phase of IR progression leads to the thresholds of irreversibility of the physiological changes as described in the Physiological Fitness Landscape (PFL) model we recently proposed [56]. However, according to the PFL model, there is potential to raise these thresholds. This phase often may lead to oncogenic carcinogenesis, hepatic cirrhosis, and atherogenesis. IR/hyperinsulinemia promotes downregulation of insulin receptors leading to IR in the brain, in particular in the hypothalamus. This is linked to impaired satiety giving rise to morbid obesity. IR in the limbic system involving the hippocampus and emotional centers/amygdala can be a precursor to AD.
Hyperinsulinemia drives lipogenesis in the liver potentiated by the influx of fatty acids coming in from visceral adipose stores of insulin resistant fat cell depots (i.e. exaggerated activation of lipolytic activity), which together promotes hepatic steatosis. This is a sine qua non manifestation of hepatic and systemic IR, and the most common cause of liver cirrhosis as well as the basis for cholesterol and lipid abnormalities that drive atherogenesis. Excessive fat accumulation in the liver overloads mitochondrial bioenergetic capacity of the electron transport systems producing ROS and oxidative stress. This drives a positive feedback binary loop of mitochondrial dysfunction and redox/oxidative stress, which may lead to oncogenic mutations and hepatic carcinoma. Hyperinsulinemia stimulates mitogenic pathways in tissue cells systemically. While insulin itself does not transform cells from a normal phenotype to the cancerous one, it promotes cell proliferation of existing cancer cells (including subclinical cancers) of tissues in the reproductive, gastrointestinal and urogenital tracts. Moreover, these mitogenic pathways promote inflammation, which in a feedforward loop of redox stress, may promote oncogenic mutations and cancer cell initiation.
When the body is in a state of chronic IR, rescue programs for cell redox damage that typically occur during the daily cycle’s sleep phase become impaired. This, together with the additional loss of various forms of adaptive flexibility, causes disturbances in the temporal balance between redox and metabolic homeostasis such as disrupted regulation of glucose and lipid output from the liver. Consequently, excessive accumulation of ectopic lipids in tissues systemically leads to cellular dysfunction mediated by redox and inflammatory stress, which ultimately promotes the pathogenesis of IR and chronic disease [57].
In some individuals, a propensity of islet dysfunction leads to declines in insulin secretion that then bring about hyperglycemia and diabetes. This is particularly amplified as the organizational precision of the body declines with age, hence desynchronization of insulin secretion with fasting/feeding and sleep/wake cycles can result in pathogenic IR.Consequently, IR loses its periodic circadian rhythm and becomes associated with relative postprandial insulinopenia and fasting hyperinsulinemia. This compromised temporal organization across the organism is mediated by impaired bioenergetics of ATP production and a reduction in the Gibbs free energy that accompanies redox stress.
Departure from a healthy dietary and consistent circadian lifestyle promotes obesityand insulin resistance and makes the individual more susceptible to premature aging and chronic diseases. Obesity is often associated with dysfunction of adipose tissue. The capacity of adipose tissue to store lipids varies, dependent on the size and number of adipocytes, which in turn vary with sex, age, fitness and genetics, etc. When this capacity is exceeded, the overflow of free fatty acid from adipose tissue into the blood, and subsequently into ectopic tissues, such as the liver and skeletal muscle leads to insulin resistance in liver and muscle [58].
ROS and reactive nitrogen species (RNS), or free radicals of oxygen- and nitrogen-containing molecules, can cause oxidative and nitrosative stress, respectively. Depending on the levels of each, it is possible for them to have either healthy or pathological effects (hormesis). Low to moderate levels of free radicals under regulated conditions promote healthy physiological effects, whereas excessive and uncontrolled levels of free radicals promote chronic disease and accelerated aging often associated with the metabolic disturbances, insulin resistance, and sometimes diabetes.An important molecular mechanism underlying the pathophysiology of oxidative stress that leads to chronic disease states of aging associated with IR and diabetes is the nonenzymatic glycation of lipids and proteins. Exposure to sugars causes glycation of lipids at phospholipid groups and proteins at lysine/arginine amino acid groups. Both cases lead to the creation of Advanced Glycation Products (AGEs).Soluble, circulating protein AGEs or AGEs formed on collagen in the extracellular space bind to Receptor for AGEs (RAGEs) on endothelial cells of blood vessels, peroneal cells, epithelial cells of the gut or reproductive tract, cardiomyocytes, pancreatic beta cells, adipocytes, and immune cells. RAGEs promote the signal transduction of AGEs, which trigger inflammatory responses and cellular dysfunction. This is also a causative link between IR and atherosclerosis and cardiovascular disease [17] since ROS promote endothelial cell dysfunction by inhibiting endothelial Nitric Oxide Synthase (eNOS) and Nitric Oxide (NO)-mediated vasodilatation [59]. This results in hypertension.
IR and hyperinsulinemia is also connected to cancer, in particular endometrial cancer, breast cancer in post-menopausal women [60] as well as renal cell, cervical, ovarian, breast, prostate, gallbladder, colorectal, gastric, esophageal, hepatocellular, non-Hodgkin’s lymphoma, and multiple myeloma [61]. Moreover, obesity at diagnosis of cancer signals a potential for a worse clinical outcome [62-64]. An integrative model [65] shows involvement of systemic insulin resistance, hyperinsulinemia, type 2 diabetes, estrogens, inflammatory cytokines, hyperlipidemia, hyperglycemia, obesity, tumorigenesis, and cancer cell proliferation. Solid evidence has been provided for a cause-and-effect relationship between hyperinsulinemia and cancer [66] with a mechanistic cellular model linking insulin and IGF-1 to cancer cell growth [67]. Insulin and IGF-1 stimulate receptor signaling through PI3K/Akt/mTOR and RAS/MAPK pathways, which promote cancer cell proliferation and/or survival, and increase tumor angiogenesis. In the setting of IR and coexisting hyperinsulinemia, hyperinsulinemia drives higher expression of Growth Hormone (GH) receptors in the liver. Another node in the insulin signaling pathway is the Ras-Raf-MAPK pathway, which plays important roles in cell regulation and mitogenesis and is responsible for tumorigenesis [68] and inflammatory effects in insulin-targeting tissues [69]. Moreover, obesity and insulin resistance/hyperinsulinemia directly promote cancer. Thus, the interwoven fabric of obesity, insulin resistance/hyperinsulinemia and inflammation provide a foundational parameter for many cancers.The upregulation of insulin signaling in cancer cells promotes cell survival, growth and replication (Figure 3).
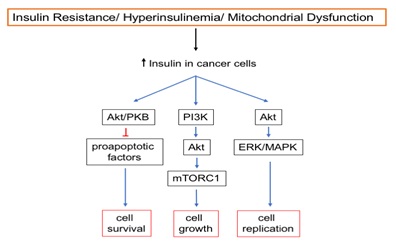 Figure 3: Insulin signaling in cancer cells promotes cell survival, cell growth, and cellular replication. Increased insulin signaling through the Akt/PKB pathway inhibits pro-apoptotic factors, resulting in increased cellular survival. Insulin signaling through the PI3K pathway results in activation of mTORC1, increasing cellular growth. Insulin signaling through the ERK/MAPK pathway upregulates cellular replication.
Figure 3: Insulin signaling in cancer cells promotes cell survival, cell growth, and cellular replication. Increased insulin signaling through the Akt/PKB pathway inhibits pro-apoptotic factors, resulting in increased cellular survival. Insulin signaling through the PI3K pathway results in activation of mTORC1, increasing cellular growth. Insulin signaling through the ERK/MAPK pathway upregulates cellular replication.
The presence of hyperinsulinemia provides extracellular stimulation that links cancer cells to the Warburg effect. This occurs when there is the preferential utilization of glucose to bioenergetically fuel cancer cell growth and proliferation via the glycolysis pathway, in the absence of oxygen consumption even when oxygen is available. Hyperinsulinemia linking cancer cells to the Warburg effect is largely underpinned by the PI3K-Akt-mTOR pathway and other Akt-mediated signaling.
Third Phase: Ectopic Fat Development
This phase generally defines thresholds of reversibility as defined in the PFL model [56] with notable exception of hepatic steatosis. The threshold of reversibility may not be achieved except, as shown by Petersen [31] that an 8kg (10%) weight loss reverses hepatic IR. A 5 % weight loss results in a 40% loss of hepatic fat. Conversely, dietary overconsumption causes adipocytes to become overstuffed with fat, which outstrips adipocyte blood supply. This then causes chronic states of hypoxia, inflammation, redox imbalance and IR triggering lipolysis. Subsequently, ectopic fat is generated and metabolic dysregulation of core self-amplifying loops across the system.
IR in adipocytes with insuppressible lipolysis and high fatty acid influx coupled with hyperinsulinemia promotes hepatic steatosis, and consequently dyslipidemia and vascular disease. These processes occurring in skeletal muscle manifest in worsening IR. Ectopic fat storage in heart muscle leads to myocardial dysfunction. Lipid droplets in the limbic system of the brain promote disturbances in emotion and cognition, while in the pancreas they cause exocrine and endocrine insufficiency and lead to GI malabsorption and T2D. Ectopic fat deposits in many epithelial tissues may result in oncogenic mutations and cancer cell initiation.
Ectopic intramyocellular lipid (IMCL) accumulation can occur even in the absence of obesity and associated spillover of fat from adipose tissue storage depots [33]. Genetic variants in enzymes that code for fatty acid oxidation in humans have shown a high incidence of insulin resistance and diabetes [70]. Individuals in the top quartile of normal glucose tolerance maintained euglycemia with a cost of hyperinsulinemia [34]. This compensatory response in turn becomes a pathogenic driver of de novo lipogenesis in the liver. It follows that hepatic de novo lipogenesis and steatosis is a hallmark of insulin resistance. Once a threshold of lipid buildup in the liver is reached, the synthesis of triglyceride and Very Low-Density Lipoprotein (VLDL) particles and their release into the circulation is accelerated. The progression of hepatic fat accumulation is responsible for triggering inflammatory liver injury, or transaminitis, which is now the most common etiology of cirrhosis and can lead to hepatic carcinoma.
In the state of type 2 diabetes, a relatively modest 10% weight loss promotes a dramatic reduction in liver fat and improvement in fasting hyperglycemia accompanying an increase in total body oxidative bioenergetics [31]. Thiazolidinedione agents and metformin, recommended as first line drug therapy for the treatment of type 2 diabetes, promote a metabolically favorable topography of body fat with a reduction in hepatic fat. Based on the lipocentric pathogenesis of insulin resistance and diabetes, it was concluded that targeting an upregulation of mitochondrial fatty acid oxidation would be another ideal pharmacotherapeutic approach. Such drugs that work by promoting the uncoupling of fatty acid oxidation to ATP production in the liver are currently in the pipeline of research and development. This approach increases the burning of acetyl CoA, rather than allowing the alternate pathway of gluconeogenesis, following delivery of excess fatty acids and glycerol to the liver due to exaggerated adipose tissue lipolysis in states of insulin resistance. The reduction in gluconeogenesis helps to normalize fasting hyperglycemia, as well as the superimposed component of relatively uninhibited hepatic glucose output by meals [71].
Mitochondrial dysfunction could also be associated with activation of the Endoplasmic Reticulum (ER) stress pathways. As mitochondrial bioenergetic capacity declines, there is an associated increase in unfolded proteins. Dysfunctional mitochondria have a reduced ability to maintain the metabolic demands of normal protein folding and are thus burdened by ER stress as this is the organelle in which protein folding takes place. ER stress and the accumulation of unfolded proteins initiate the unfolded protein response which activates branches of inflammatory (NFκB and JNK) and oxidative stress pathways that all work to aggressively promote insulin resistance. Mitochondrial dysfunction thus represents an impaired capacity to transform available energy substrates into the biological currency of ATP in a manner sufficient to maintain cellular homeostasis.Some degree of mitochondrial dysfunction is present in most individuals who are predisposed to insulin resistance. Mitochondrial dysfunction will fundamentally decrease bioenergetic metabolism, altering the balance between substrate availability and oxidative capacity.The interconnections between mitochondrial dysfunction and insulin resistance are complex and varied. Mitochondrial function and insulin signaling are also influenced by numerous hormonal, neural, circadian and metabolic inputs.
The brain’s high demand for energy makes it vulnerable to the accumulation of dysfunctional mitochondria, which is associated with neurodegenerative disorders including Alzheimer’s disease. Defective mitochondria generate ROS, which can eventually lead to neuronal death [72-74]. Mitophagy serves to preserve the nervous system’s physiology, and can drive the pathogenesis of Alzheimer’s and other neurodegenerative diseases when it fails [75,76]. The energy deficit and excess ROS generated by dysfunctional mitochondria promote insulin resistance in the brain, and ultimately amyloid beta accumulation with clinical signs of Alzheimer’s [77-79]. Obesity is an established risk factor for Alzheimer’s disease with ectopic fat accumulation in the brain causing reduced insulin signaling and accelerated cognitive decline. Accumulation of ceramides has been demonstrated to cause brain mitochondrial dysfunction. Insulin signaling in the brain regulates the synaptic release and reuptake of various neurotransmitters in areas such as the hypothalamus and hippocampus, which mediate satiety as well as learning and memory [80-83]. In fact, disrupting insulin signaling in the brain may lead to impaired spatial memory and anxiety-like behaviors.
There is a bidirectional relationship between accelerated cognitive decline and Alzheimer's disease with insulin resistance and diabetes type 2. Between the ages of 60 and 74, the age range when accelerated cognitive decline and Alzheimer's disease incidence precipitously increases, an estimated 67% of the United States population is diabetic or pre-diabetic. The cerebral cortex and hippocampus, two regions of the brain that are highly sensitive to insulin, require both glucose and insulin for high level cognitive functions such as learning and memory consolidation.
Fourth Phase: Stress Resistance Programs
In this final stage of IR progression, redox stress resilience programs including autophagy/mitophagy and apoptosis are activated. Clinical manifestations can be seen to pass the threshold of reversibility. In the liver, the presentation of this stage of IR involves a cirrhotic shrunken liver. In skeletal muscle, sarcopenia and muscle wasting is evident and a related sign of mortality is how fast an older person can walk. The heart tissue presents with dilated cardiomyopathy. The loss in cell volume of the brain’s hippocampus causes accelerated cognitive decline. Finally, IR effects in the pancreas lead to pancreatic insufficiency.
The hormone leptin also plays a role in responses to stress. Leptin exerts a satiety effect through regulation of neuropeptides, by signaling through the leptin receptor, found on neurons in the arcuate nucleus in the hypothalamus [84]. Overall, leptin acts to reduce food intake. During leptin resistance seen in states of obesity, the positive energy balance is further potentiated. Perhaps independently of leptin's effect on the circadian rhythm of the hypothalamic pituitary adrenal, gonadal and thyroid axes are additional effects on neuroendocrine function. Moreover, the hypothalamic pituitary growth hormone axis is also regulated by leptin [85]. It follows that leptin resistance is associated with a blunted growth hormone response [86,87]. Thus, growth hormone-related effects of leptin resistance are clinically most relevant to an underlying state of insulin resistance and obesity as well as vitamin D deficiency.
Vitamin D promotes osteocalcin gene expression that enhances insulin sensitivity, thus improving insulin resistance, obesity, glucose intolerance and so on. Leptin regulates the hypothalamic pituitary adrenal axis by inhibiting cortisol release from adrenocortical cells [88,89]. There is crosstalk between leptin and cortisol centrally in the hypothalamic paraventricular nucleus, where leptin opposes cortisol feedback suppression of CRH mediated by endocannabinoid biosynthesis and release. Thus, leptin disinhibits the feedback suppression of cortisol, potentiating the CRH driven stress response. Hence, cortisol and leptin have antagonistic effects on the neuronal stress response and associated energy homeostasis centrally at the level of the hypothalamic paraventricular nucleus. Accordingly, leptin resistance attenuates the cortisol stress response. This relationship is interesting because while leptin resistance promotes obesity, insulin resistance and metabolic disease states downstream of these parameters, it counterbalances the toxic metabolic effects of the prolonged HPA stress response.
In the case of psychogenic stressors, imagined stress when acute may be motivating; hence adaptive stressors activating the sympathetic nervous system and hypothalamic pituitary stress response may be lifesaving in the case of a real physical threat. However, psychogenic stressors are often pathologically prolonged in modern society, thus overextending the adaptive nature of the stress response leading to pathology or disease.
Psychogenic stressors may take many forms. Being the caretaker of a sick and elderly parent, or other loved one, ranks among one of the most stressful circumstances in life. Loss of physical health is also a significant psychogenic stressor; importantly, this relationship is bidirectional. Perceived stress worsens physical health, and mediated by inflammatory protein cytokines, physical health decline reduces the threshold for tolerance of external stressors. Social stress ranks very high in the pathological potential of psychogenic stressors. Social stressors may include chronic relationship discord or social isolation - isolated from love, networking and interactions with friends and family. Financial stress is common and may be an absolute stress due to unemployment and lack of financial resources. Work stress fundamentally is rooted in failure or fear of failure of performing the work responsibilities to expectations including personal expectations. Work stress may be importantly rooted in the lack of empathy, compassion or other motivation required to put personal talents to work.
Nutrient disturbances also contribute to pathology and may be in the form of three pathological patterns: nutrient depletion; energy excess; and dietary toxicants, the pollution of diet with greater than 80,000 processing chemicals introduced in the 1970s. It is worthwhile in making the above distinctions of psychogenic stressors and nutrient disturbances as parameters in order to discern them as targets of upstream interventions to prolonged and pathogenic parameters of the neuro-endocrine stress responses. As described above, these become parameters for insulin resistance and subsequent disease states including not only type 2 diabetes but chronic disease states of accelerated cognitive decline, Alzheimer's disease, cardiovascular disease and cancers, which should all be recognized as metabolic disease states.
Stress is any situation that may disturb the equilibrium of a living organism and its environment. This equilibrium is asymmetrical, an adaptive homeostasis of the organism in the setting of its environment. The responses of the organism to the stress are allostatic responses to maintain homeostasis. These allostatic neuroendocrine responses include the secretion of Corticotropin Releasing Hormone (CRH) from the paraventricular nucleus of the hypothalamus, enhanced pituitary hormone secretion, growth hormone, prolactin and vasopressin, adrenal gland glucocorticoids, and the sympathetic nervous system release of catecholamines. These stress responses are considered necessary for survival in the classical fight or flight setting to adaptively increase mobilization of energy sources, optimized immune function as well as wound healing.
Growth hormone is increased during acute physical stress and antagonizes insulin signaling, hence promoting energy mobilization and allowing availability of energy sources to the body systemically. Interestingly, chronic psychological stress is not usually associated with a growth hormone response. Importantly, it should be highlighted that the purpose of the acute stress response is to promote the calibration of limited energy resources to the fight or flight response intended to keep the organism alive from an evolutionary perspective. Accordingly, it calibrates these resources away from energy expensive processes such as reproduction.
Fundamentally, functions of cortisol and growth hormone antagonize insulin signaling, which is important to the overall response of promoting glucose primarily to the brain, and glucose and fatty acid delivery to the heart and skeletal muscles. This allows the appropriate preferred energy source to their respective tissues to optimize the fight or flight response. In addition to the hormonal component of the neuro-endocrine response, the catecholamine response is inextricably part of the fabric of this stress response. Catecholamines importantly contribute to nutrient delivery as well as other independent effects on sharpening cognitions, sight, hearing and other senses that put the body in a state of hair-trigger alert. Some of the effects of catecholamine that mediate the stress and hence fight or flight responses include hemodynamics such as activating the renin-angiotensin-aldosterone system promoting sodium retention from the kidney, the release of Antidiuretic Hormone (ADH) or vasopressin, promoting retention of free water, and increased cardiac output, all of which promote the adequate flow of blood carrying nutrients and oxygen to peripheral tissues.
Catecholamines as neurotransmitters of the sympathetic nervous system reduce intestinal motility and increase cutaneous vasoconstriction. This promotes gluconeogenesis, the production of glucose from other nutrient precursors such as lactate, amino acids and glycerol in a variety of tissues, most importantly the liver. Through beta receptors, catecholamines enhance bronchodilation for deep breathing, allowing oxygenation of tissues during this high metabolic demand state. When the stress response is excessive or prolonged, a state of allostatic load ensues. In this state, the allostatic responsiveness exhibits a loss in resilience of the body against the external perturbations. Excessive or prolonged stress response may be rooted in epigenetic factors, that is, environmental exposures as well as genetic predispositions for abnormal perceptions of stress. It may also be rooted in psychogenic stress, whether real or imagined.
Conclusion
The inextricable intertwining of disturbed redox homeostasis, inflammation and impaired bioenergetics appear to be fundamentally linked to the chronic diseases of aging, including CVD, dementias and cancers. These manifestations of advanced and often premature biological aging are largely mediated by pathogenic parameters of: disturbed microbiota, a chronic exaggerated stress response, and dis-synchronous circadian metabolism and physiology within and between tissues. All of these factors are also inseparably entangled.
- Insulin resistance occurs under both healthy and pathologic conditions. In healthy tissue insulin resistance is maintained under cyclical circadian patterns that are adaptive and maintain cell stress resilience programs. Conversely, insulin resistance becomes pathological when not following cyclical circadian patterns
- Parameters of insulin resistance include prolonged psychogenic stressors, poor diet quantity and quality, disrupted circadian behaviors, and altered gut microbiota
- The Respiratory Quotient (RQ) is a clinically useful tool to measure mitochondrial inflexibility, which is indicative of insulin resistant states
- Increased levels of ROS and AGEs generate inflammation and are associated with insulin resistance
- Insulin resistance affects the different metabolic tissues of the body (i.e., liver, skeletal and cardiac muscles, and adipose tissues) in unique ways
- There is a bidirectional self-amplifying relationship between insulin resistance and mitochondrial dysfunction, among 4 other core self-amplifying binary loops, where either can drive the other
- High levels of free radicals and oxidative stress are associated with insulin resistance, while low levels promote healthy physiological effects
- Insulin resistance contributes to the pathogenesis of cardiovascular disease
- Insulin resistance contributes to the pathogenesis of cancer via the Warburg effect. This effect can be described as the inability of native host cells to efficiently utilize available oxygen to meet their bioenergetic needs. Thus, cancer cells can outcompete them for available resources using glycolytic metabolism
- Insulin resistance can contribute to the pathogenesis of Alzheimer’s disease and accelerated cognitive decline
References
- Henquin JC, Gembal M, Detimary P, Gao ZY, Warnotte C, et al. (1994) Multisite control of insulin release by glucose. Diabete Metab 20: 132-137.
- Newgard CB, McGarry JD (1995) Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu Rev Biochem 64: 689-719.
- Cheng Z, Tseng Y, White MF (2010) Insulin signaling meets mitochondria in metabolism. Trends Endocrinol Metab 21: 589-598.
- Nisr RB, Affourtit C (2014) Insulin acutely improves mitochondrial function of rat and human skeletal muscle by increasing coupling efficiency of oxidative phosphorylation. Biochim Biophys Acta 1837: 270-276.
- Crosby P, Hamnett R, Putker M, Hoyle NP, Reed M, et al. (2019) Insulin/IGF-1 drives PERIOD synthesis to entrain circadian rhythms with feeding time. Cell 14: 173-194.
- Hales CN, Randle PJ (1963) Effects of low-carbohydrate diet and diabetes mellitus on plasma concentrations of glucose, non-esterified fatty acid, and insulin during oral glucose-tolerance tests. Lancet 1: 790-794.
- Defronzo RA (2009) Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes 58: 773-795.
- Salans LB, Knittle JL, Hirsch J (1968) The role of adipose cell size and adipose tissue insulin sensitivity in the carbohydrate intolerance of human obesity. J Clin Invest 47: 153-165.
- Krotkiewski M, Björntorp P, Sjöström L, Smith U (1983) Impact of obesity on metabolism in men and women. Importance of regional adipose tissue distribution. J Clin Invest 72: 1150-1162.
- Weyer C, Foley JE, Bogardus C, Tataranni PA, Pratley RE (2000) Enlarged subcutaneous abdominal adipocyte size, but not obesity itself, predicts type II diabetes independent of insulin resistance. Diabetologia 43: 1498-1506.
- Trayhurn P, Alomar SY (2015) Oxygen deprivation and the cellular response to hypoxia in adipocytes - perspectives on white and brown adipose tissues in obesity. Front Endocrinol 6: 19.
- Klöting N, Blüher M (2014) Adipocyte dysfunction, inflammation and metabolic syndrome. Rev Endocr Metab Disord 15: 277-287.
- de Heredia FP, Gómez-Martínez S, Marcos A (2012) Obesity, inflammation and the immune system. Proc Nutr Soc 71: 332-338.
- Ye JM, Tid-Ang J, Turner N, Zeng X-Y, Li H-Y, et al. (2011) PPARδ agonists have opposing effects on insulin resistance in high fat-fed rats and mice due to different metabolic responses in muscle. Br J Pharmacol 163: 556-566.
- Matsuzaka T, Shimano H (2011) Molecular mechanisms involved in hepatic steatosis and insulin resistance. J Diabetes Investig 2: 170-175.
- Kashyap SR, Defronzo RA (2007) The insulin resistance syndrome: Physiological considerations. Diab Vasc Dis Res 4: 13-19.
- Di Pino A, DeFronzo RA (2019) Insulin resistance and atherosclerosis: Implications for insulin-sensitizing agents. Endocr Rev 40: 1447-1467.
- Petersen KF, Shulman GI (2006) Etiology of insulin resistance. Am J Med 119: 10-16.
- Samuel VT, Petersen KF, Shulman GI (2010) Lipid-induced insulin resistance: unravelling the mechanism. Lancet 375: 2267-2277.
- Perry RJ, Samuel VT, Petersen KF, Shulman GI (2014) The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature 510: 84-91.
- Szendroedi J, Roden M (2008) Mitochondrial fitness and insulin sensitivity in humans. Diabetologia 51: 2155-2167.
- Burkart AM, Tan K, Warren L, Iovino S, Hughes KJ, et al. (2016) Insulin Resistance in Human iPS Cells Reduces Mitochondrial Size and Function. Sci Rep 6: 22788.
- Kim JY, Park KJ, Hwang JY, Kim GH, Lee DY, et al. (2017) Activating transcription factor 3 is a target molecule linking hepatic steatosis to impaired glucose homeostasis. J Hepatol 67: 349-359.
- Karakelides H, Asmann YW, Bigelow ML, Short KR, Dhatariya K, et al. (2007) Effect of insulin deprivation on muscle mitochondrial ATP production and gene transcript levels in type 1 diabetic subjects. Diabetes 56: 2683-2689.
- Szendroedi J, Schmid AI, Chmelik M, Toth C, Brehm A, et al. (2007) Muscle mitochondrial ATP synthesis and glucose transport/phosphorylation in type 2 diabetes. PLoS Med 4: 154.
- Schmid AI, Szendroedi J, Chmelik M, Krssák M, Moser E, et al. (2011) Liver ATP synthesis is lower and relates to insulin sensitivity in patients with type 2 diabetes. Diabetes Care 34: 448-453.
- Mogensen M, Sahlin K, Fernström M, Glintborg D, Vind BF, et al. (2007) Mitochondrial respiration is decreased in skeletal muscle of patients with type 2 diabetes. Diabetes 56: 1592-1599.
- Pinti MV, Fink GK, Hathaway QA, Durr AJ, Kunovac A, et al. (2019) Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am J Physiol Endocrinol Metab 316: 268-285.
- DeFronzo RA, Ferrannini E (1991) Insulin resistance. A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidemia, and atherosclerotic cardiovascular disease. Diabetes Care 14: 173-194.
- Reaven GM (2005) Why Syndrome X? From Harold Himsworth to the insulin resistance syndrome. Cell Metab 1: 9-14.
- Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, et al. (2005) Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes 54: 603-608.
- Befroy DE, Petersen KF, Dufour S, Mason GF, Graaf RA, et al. (2007). Impaired mitochondrial substrate oxidation in muscle of insulin-resistant offspring of type 2 diabetic patients. Diabetes 56: 1376-1381.
- Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, et al. (2003) Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science 300: 1140-1142.
- Petersen KF, Dufour S, Savage DB, Bilz S, Solomon G, et al. (2007) The role of skeletal muscle insulin resistance in the pathogenesis of the metabolic syndrome. Proc Natl Acad Sci USA 104: 12587-12594.
- Petersen KF, Oral EA, Dufour S, Befroy D, Ariyan C, et al. (2002) Leptin reverses insulin resistance and hepatic steatosis in patients with severe lipodystrophy. J Clin Invest 109: 1345-1350.
- Bruls YM, de Ligt M, Lindenboom L, Phielix E, Havekes B, et al. (2019) Carnitine supplementation improves metabolic flexibility and skeletal muscle acetylcarnitine formation in volunteers with impaired glucose tolerance: A randomised controlled trial. EBioMedicine 49: 318-330.
- Summers SA (2018) Could ceramides become the new cholesterol? Cell Metab 27: 276-280.
- Havulinna AS, Sysi-Aho M, Hilvo M, Kauhanen D, Hurme R, et al. (2016) Circulating ceramides predict cardiovascular outcomes in the population-based FINRISK 2002 cohort. Arterioscler Thromb Vasc Biol 36: 2424-2430.
- Neeland IJ, Singh S, McGuire DK, Vega GL, Roddy T, et al. (2018) Relation of plasma ceramides to visceral adiposity, insulin resistance and the development of type 2 diabetes mellitus: the Dallas Heart Study. Diabetologia 61: 2570-2579.
- Petrocelli JJ, McKenzie AI, Mahmassani ZS, Reidy PT, Stoddard GJ, et al. (2020). Ceramide biomarkers predictive of cardiovascular disease risk increase in healthy older adults after bed rest. J Gerontol A Biol Sci Med Sci 75: 1663-1670.
- Poss AM, Maschek JA, Cox JE, Hauner BJ, Hopkins PN, et al. (2020) Machine learning reveals serum sphingolipids as cholesterol-independent biomarkers of coronary artery disease. J Clin Invest 130: 1363-1376.
- Cogolludo A, Frazziano G, Cobeño L, Moreno L, Lodi F, et al. (2006) Role of reactive oxygen species in Kv channel inhibition and vasoconstriction induced by TP receptor activation in rat pulmonary arteries. Ann N Y Acad Sci 1091: 41-51.
- Vizcaino FP, Cogolludo A, Moreno L (2010) Reactive oxygen species signaling in pulmonary vascular smooth muscle. Respir Physiol Neurobiol 174: 212-220.
- Montezano AC, Lis MD, Tsiropoulou S, Harvey A, Briones AM, et al. (2015) Oxidative stress and human hypertension: vascular mechanisms, biomarkers, and novel therapies. Can J Cardiol 31: 631-641.
- Cogolludo A, Villamor E, Vizcaino FP, Moreno L (2019) Ceramide and Regulation of Vascular Tone. Int J Mol Sci 20: 411.
- Hojjati MR, Li Z, Zhou H, Tang S, Huan C, et al. (2005) Effect of myriocin on plasma sphingolipid metabolism and atherosclerosis in apoE-deficient mice. J Biol Chem 280: 10284-10289.
- Park TS, Panek RL, Rekhter MD, Mueller SB, Rosebury WS, et al. (2006) Modulation of lipoprotein metabolism by inhibition of sphingomyelin synthesis in ApoE knockout mice. Atherosclerosis 189: 264-272.
- Glaros EN, Kim WS, Wu BJ, Suarna C, Quinn CM, et al. (2007) Inhibition of atherosclerosis by the serine palmitoyl transferase inhibitor myriocin is associated with reduced plasma glycosphingolipid concentration. Biochem Pharmacol 73: 1340-1346.
- Bharath LP, Ruan T, Li Y, Ravindran A, Wan X, et al. (2015) Ceramide-initiated protein phosphatase 2A activation contributes to arterial dysfunction in vivo. Diabetes 64: 3914-3926.
- Chaurasia B, Tippetts TS, Mayoral Monibas R, Liu J, Li Y, et al. (2019) Targeting a ceramide double bond improves insulin resistance and hepatic steatosis. Science 365: 386-392.
- Field BC, Gordillo R, Scherer PE (2020) The role of ceramides in diabetes and cardiovascular disease regulation of ceramides by adipokines. Front Endocrinol (Lausanne) 11: 569250.
- Brooks IV, Sounier R, Rochaix P, Bellot G, Fortier M, et al. (2017) Structural insights into adiponectin receptors suggest ceramidase activity. Nature 544: 120-123.
- Kim JY, Wall E, Laplante M, Azzara A, Trujillo ME, et al. (2007) Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest 117: 2621-2637.
- Smith RL, Soeters MR, Wüst RCI, Houtkooper RH (2018) Metabolic flexibility as an adaptation to energy resources and requirements in health and disease. Endocr Rev 39: 489-517.
- Galgani JE, Jonge L, Most MM, Bray GA, Smith SR (2010) Effect of a 3-day high-fat feeding period on carbohydrate balance and ad-libitum energy intake in humans. Int J Obes (Lond) 34: 886-891.
- Fertig BJ, Chopra D and Tuszynski JA, (2021). Metabolism, microbiota-host interactions, aging and stress response: diagnostic and therapeutic applications of the Physiological Fitness Landscape. J Altern Complement Integr Med 7: 211.
- Bays H, Mandarino L, DeFronzo RA (2004) Role of the adipocyte, free fatty acids, and ectopic fat in pathogenesis of type 2 diabetes mellitus: peroxisomal proliferator-activated receptor agonists provide a rational therapeutic approach. J Clin Endocrinol Metab 89: 463-478.
- Mittendorfer B (2011) Origins of metabolic complications in obesity: adipose tissue and free fatty acid trafficking. Curr Opin Clin Nutr Metab Care 14: 535-541.
- Cersosimo E, DeFronzo RA (2006) Insulin resistance and endothelial dysfunction: The road map to cardiovascular diseases. Diabetes Metab Res Rev 22: 423-436.
- Morimoto LM, White E, Chen Z, Chlebowski RT, Hays J, et al. (2002) Obesity, body size, and risk of postmenopausal breast cancer: the Women's Health Initiative (United States). Cancer Causes Control 13: 741-751.
- Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ (2003). Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med 348: 1625-1638.
- Sinicrope FA, Foster NR, Sargent DJ, O'Connell MJ, Rankin C (2010) Obesity is an independent prognostic variable in colon cancer survivors. Clin Cancer Res 16: 1884-1893.
- Conroy SM, Maskarinec G, Wilkens LR, White KK, Henderson BE, et al. (2011) Obesity and breast cancer survival in ethnically diverse postmenopausal women: The Multiethnic Cohort Study. Breast Cancer Res Treat 129: 565-574.
- Orgel E, Genkinger JM, Aggarwal D, Sung L, Nieder M, et al. (2016) Association of body mass index and survival in pediatric leukemia: a meta-analysis. Am J Clin Nutr 103: 808-817.
- Roith DL, Baserga R, Helman L, Roberts CT (1995) Insulin-like growth factors and cancer. Ann Intern Med 122: 54-59.
- Roith DL, Roberts CT (2003) The insulin-like growth factor system and cancer. Cancer Lett 195: 127-137.
- Hursting SD, Berger NA (2010). Energy balance, host-related factors, and cancer progression. J Clin Oncol 28: 4058-4065.
- Michael JV, Wurtzel JG, Goldfinger LE (2016) Regulation of H-Ras-driven MAPK signaling, transformation and tumorigenesis, but not PI3K signaling and tumor progression, by plasma membrane microdomains. Oncogenesis 5: 228.
- Nandipati KC, Subramanian S, Agrawal DK (2017) Protein kinases: mechanisms and downstream targets in inflammation-mediated obesity and insulin resistance. Mol Cell Biochem 426: 27-45.
- Knowles JW, Xie W, Zhang Z, et al. (2015) Identification and validation of N-acetyltransferase 2 as an insulin sensitivity gene. J Clin Invest 126: 403.
- Samuel VT, Shulman GI (2016) The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126: 12-22.
- McLennan HR, DegliEsposti M (2000) The contribution of mitochondrial respiratory complexes to the production of reactive oxygen species. J Bioenerg Biomembr 32: 153-162.
- Ying W (2008) NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal 10: 179-206.
- Kauser H, Chopra D, Mukherjee S, Mohan P (2018) Pharmacoepidemiological Observational Study of Antimicrobial Use in Outpatients of Ophthalmology Department in North Indian Population. J Pharm Bioallied Sci 10: 72-76.
- Chakravorty A, Jetto CT, Manjithaya R (2019) Dysfunctional mitochondria and mitophagy as drivers of Alzheimer's disease pathogenesis. Front Aging Neurosci 11: 311.
- Cai Q, Jeong YY (2020) Mitophagy in Alzheimer's disease and other age-related neurodegenerative diseases. Cells 9: 150.
- Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, et al. (2010) Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer's disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 31: 224-243.
- Camandola S, Plick N, Mattson MP (2019) Impact of coffee and cacao purine metabolites on neuroplasticity and neurodegenerative disease. Neurochem Res 44: 214-227.
- Acosta JB, Nieto GG, Rodríguez NR, et al. (2020). Insulin resistance at the crossroad of Alzheimer disease pathology: A review. Front Endocrinol (Lausanne) 11: 560375.
- Saltiel AR, Kahn CR (2001) Insulin signalling and the regulation of glucose and lipid metabolism. Nature 414: 799-806.
- Zhao WQ, Chen H, Quon MJ, Alkon DL (2004) Insulin and the insulin receptor in experimental models of learning and memory. Eur J Pharmacol 490: 71-81.
- Biessels GJ, Kappelle LJ, Utrecht Diabetic Encephalopathy Study Group (2005) Increased risk of Alzheimer's disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans 33: 1041-1044.
- Schwartz MW, Porte D (2005) Diabetes, obesity, and the brain. Science 307: 375-379.
- Ahima RS, Qi Y, Singhal NS (2006) Adipokines that link obesity and diabetes to the hypothalamus. Prog Brain Res 153: 155-174.
- Khan SM, Hamnvik OP, Brinkoetter M, Mantzoros CS (2012) Leptin as a modulator of neuroendocrine function in humans. Yonsei Med J 53: 671-679.
- Marinis LD, Bianchi A, Mancini A, Gentilella R, Perrelli M, et al. (2004) Growth hormone secretion and leptin in morbid obesity before and after biliopancreatic diversion: relationships with insulin and body composition. J Clin Endocrinol Metab 89: 174-180.
- Myers MG, Heymsfield SB, Haft C, Kahn BB, Laughlin M, et al. (2012) Challenges and opportunities of defining clinical leptin resistance. Cell Metab 15: 150-156.
- Bornstein SR, Uhlmann K, Haidan A, Bornstein ME, Scherbaum WA (1997) Evidence for a novel peripheral action of leptin as a metabolic signal to the adrenal gland: leptin inhibits cortisol release directly. Diabetes 46: 1235-1238.
- Szücs N, Varga I, Jakab C, Patócs A, Gláz E, et al. (2001) Leptin inhibits cortisol and corticosterone secretion in pathologic human adrenocortical cells. Pituitary 4: 71-77.
Citation: Fertig BJ, Chopra D, Tuszynski JA (2022) The Four Stages of Insulin Resistance Induced Chronic Diseases of Aging. J Altern Complement Integr Med 8: 232.
Copyright: © 2022 Brian J Fertig, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.