The Pharmacologic Rationale Supporting the Potential Use of Vepoloxamer for the Treatment of Acute Chest Syndrome in Patients with Sickle Cell Disease
*Corresponding Author(s):
Martin EmanueleMast Therapeutics Inc, Altai Branch, Valley Centre, San Diego, CA, United States
Tel:+1 8585520866,
Email:memanuele@mastthera.com
Abstract
Sickle Cell Disease (SCD) is an autosomal recessive disorder caused by a single amino acid substitution in the Hemoglobin (Hb) β chain which causes it to polymerize in the deoxy state. Acute Chest Syndrome (ACS) with hypoxemic respiratory failure is a common cause of death in adult SCD subjects. Recent research has revealed how multiple disease processes including inappropriate cellular adhesion, hemolysis, dysfunctional coagulation, inflammation, and ischemia and tissue injury are involved in ACS pathophysiology. In this review article, we discuss how the activity of the highly purified non-ionic amphiphilic triblock copolymer vepoloxamer in the repair of damaged membranes and hindrance of inappropriate adhesive interactions and activation events potentially has utility in ACS, by virtue of its capacity for affecting multiple aspects of the pathophysiology.
Keywords
INTRODUCTION
Sickle Cell Disease (SCD) is an autosomal recessive disorder caused by a single amino acid substitution in the Hemoglobin (Hb) β chain which causes it to polymerize in the deoxy state. The Hb polymerization results in Red Blood Cells (RBC) which are rheologically impaired and become entrapped in the microvasculature resulting in tissue ischemia [1]. Vaso-occlusive Crisis (VOC) is the hallmark of the disease and consists of severe episodic pain in the back, chest, abdomen, and joints. A frequent complication of VOC is a pulmonary illness known as Acute Chest Syndrome (ACS) typified by new pulmonary infiltrate on chest X-ray along with leukocytosis, fever, cough, chest pain, and other pulmonary symptoms [2]. The prospective cooperative study of sickle cell disease observed an incident rate of 12.8 ACS episodes /100 patient-years for homozygous SCD [3]. ACS with hypoxemic respiratory failure is a common cause of death in adult SCD subjects [4].
The origin of ACS has been associated with a wide variety of pathological processes including infection, fat embolus, and lung tissue infarction [5]. Regardless of the origin, pulmonary vaso-occlusion is likely the common pathophysiological pathway to its clinical manifestation [6]. Underlying the vaso-occlusion is the polymerization of Hemoglobin S (HbS) under hypoxic conditions, and broad endothelial dysfunction with elevated expression of multiple cell adhesion molecules contributing to vascular leukocyte recruitment, inflammation, and the activation of coagulation with thrombosis [7]. Hemolysis and the resulting increase in cell-free plasma Hb further exacerbates the endothelial dysfunction as Nitric Oxide (NO) bioavailability is reduced from the NO scavenging by cell-free heme [8], a compound which also activates innate immunity following binding of endothelial Toll-like receptor 4 [9].
With the notable exception of the polymerization of HbS, vepoloxamer has been shown to affect many of the pathological processes active in ACS, either prophylactically by interfering with activation events which drive pathogenesis, or through palliative actions against processes associated with progressive clinical pathology. Examples include: inhibiting RBC aggregation thus lowering whole blood viscosity, stabilizing of RBC membranes thereby reducing hemolysis and release of free heme, and preventing adhesive interactions between blood cells and activated endothelium thereby inhibiting occlusion, inflammation, thrombosis, and ischemic tissue damage. This publication summarizes research studies investigating the impact of vepoloxamer on those processes that are relevant to ACS pathophysiology. In doing so, we provide information on the potential mechanisms through which vepoloxamer may have utility in the treatment of ACS, a clinical conditions for which there are otherwise few therapeutic options.
VEPOLOXAMER
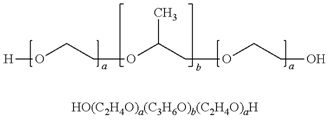
Vepoloxamer is a highly purified, non-ionic amphiphilic triblock copolymer of Polyoxypropylene (POP) flanked on both sides by Polyoxyethylene (POE) [10] (see figure 1). Its basic structure confers surface active properties that enable the modulation of a cell membrane’s biophysical properties including its stability, hydration, repair, flexibility, and adhesive properties, all of which serve crucial roles in biological responses [11]. Vepoloxamer is currently under study in a Phase III trial to evaluate whether it can reduce the duration of VOC (i.e., the EPIC study: Evaluation of Purified Poloxamer 188 In Crisis. Clinical Trials.gov Identifier: NCT01737814). The study also will evaluate whether vepoloxamer can reduce the frequency of re-hospitalizations following crisis, and the incidence of ACS during VOC.
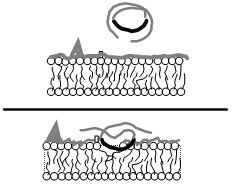
VEPOLOXAMER’S ACTIVITIES AND THEIR POTENTIAL UTILITY IN THE TREATMENT OF ACS
The following is a survey of vepoloxamer’s in vitro activities which may be relevant to ACS’s pathophysiology.
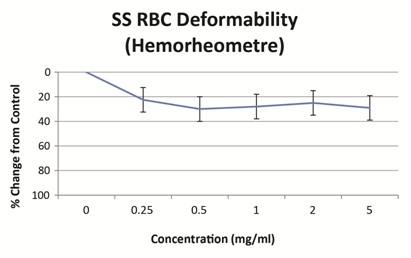
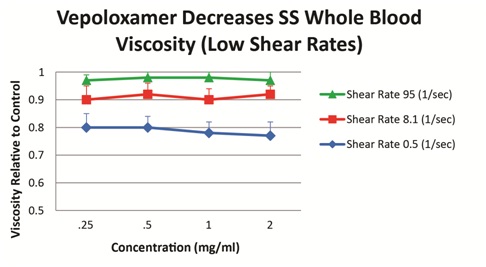
Vepoloxamer has been shown to exert concentration-dependent effects on whole blood viscosity both in healthy and SCD patients. When added to blood obtained from steady-state SCD patients, vepoloxamer at concentrations between 0.25mg/ml-2.0mg/ml showed little or no effect on whole blood viscosity at higher shear rates, while at lower shear conditions, it caused in a 20-23% reduction in viscosity (Figure 5). These observations suggest vepoloxamer may help normalize elevated viscosity as may be occurring during ACS.
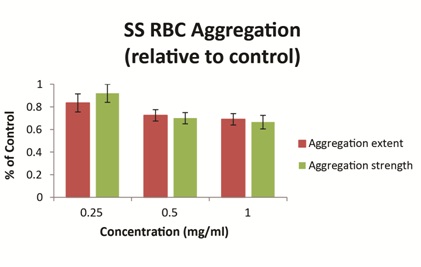
Ex vivo studies using blood obtained from patients with SCD have demonstrated that vepoloxamer exerts a concentration-dependent effect on the strength of dextran-induced sRBC aggregation. At clinically relevant concentrations (0.5mg/ml-1.0mg/ml), vepoloxamer reduces both the extent of sRBC aggregation and the strength of RBC aggregates (measured by the shear force required to break up aggregates) by approximately 30% relative to phosphate buffered saline control (Figure 6). Other methods of RBC aggregation (erythrocyte sedimentation rate and microscopic aggregation index yielded similar dose-dependent effects for vepoloxamer (data not shown). The results demonstrate that vepoloxamer causes robust, concentration-dependent decreases of both the extent and strength of RBC aggregation and suggest that vepoloxamer may reduce RBC aggregation during ACS.
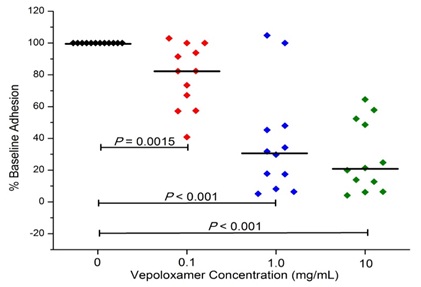
In studies to date, vepoloxamer has been shown to exhibit potent activity in inhibiting VCAM-mediated adhesion. Hsu and colleagues [37] compared the effect of vepoloxamer to the blocking ability of antibodies specific to VCAM, CD-29 and CD49 on the LPS-induced adhesion of RBC and leukocytes in the cremastor microcirculation of transgenic sickle mice. In control animals, LPS resulted in a 27% reduction in the number of perfused capillaries (which was inversely proportional to the number of adherent RBC and leukocytes). Pre-treatment with anti-VCAM, completely inhibited the adverse effects of LPS. Treatment with vepoloxamer yielded nearly identical inhibitory effects. The effects of vepoloxamer on VCAM-mediated adhesion have also been studied using freshly collected blood from patients with SCD using a microfluidics assay. In those studies, a potent, concentrated related reduction in VCAM-mediated adhesion was observed. At clinically relevant concentrations (0.1-1.0mg/ml) vepoloxamer induced a greater than a 60% reduction in adhesion compared to negative control (Figure 7).
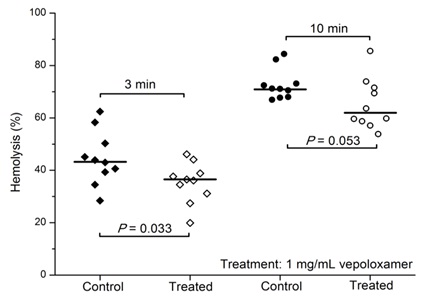
Vepoloxamer and its unpurified parent compound (poloxamer 188) are well documented for their membrane sealant and stabilizing properties in a wide variety of cells to a wide variety of mechanical and chemical challenges [12,15,26]. Baek demonstrated enhanced survival of cultured RBC in the presence of poloxamer 188 suggesting it decreased the hydrodynamic stress induced membrane injury experienced under culture conditions [41]. Recently, the effect of vepoloxamer on shear-induced hemolysis of sRBC was investigated by Hines and colleagues [38]. When subject to a physiologically relevant shear rate, vepoloxamer at therapeutically-relevant concentrations, showed a modest but statistically significant reduction in hemolysis (Figure 8).
At pharmacologically active concentrations, vepoloxamer does not exhibit anticoagulant activity and does not alter platelet function when studied in standard coagulation and platelet aggregation assays [26]. However, it does inhibit shear-induced whole blood platelet aggregation [45] and inhibits the procoagulant actions of circulating microparticles in sickle cell anemia patients [46]. These latter observations are consistent with reports describing potent antithrombotic activity following treatment [47] and collectively suggest that vepoloxamer may moderate the dysfunctional coagulation and thrombosis occurring in ACS.
EFFECT OF VEPOLOXAMER IN EXPERIMENTAL ACS
Vepoloxamer was studied in a human transgenic murine model of ACS (i.e., HbS Tg). Mice expressing human HbS at levels of either 60% or 100% were treated with a single intravenous 400mg/kg dose of vepoloxamer (n=20) or a similar volume of saline (control) (n=20) and exposed to hypoxia (5% O2). Following hypoxia, the number of sickled RBCs in the peripheral blood increased with time, and then sharply decreased before the onset of ACS symptoms. Fourteen of twenty control (60 and 100% HbS) Tg mice died within 60 minutes. In marked contrast, fifteen of twenty vepoloxamer-treated (60 and 100% HbS) Tg mice survived for 60 minutes (i.e., the total duration of the experiment).
Lung pathology was evaluated in 13 control and 13 vepoloxamer-treated HbS Tg mice, and the findings are summarized in table 1. Thrombosis was detected in all 13 control mice and was considered of a severe nature in 10 of 13 animals, and non-severe in the other three. Thrombosis was only observed in 2 of 13 vepoloxamer-treated mice and both instances it was considered to be non-severe. Nine of 13 control mice showed both RBC infiltration and partial damage of the intervening septa wall, while none of the vepoloxamer-treated mice had such changes. Leakage of RBCs into alveolar space was found in 8 of 13 control mice, while it was not present in any of the vepoloxamer-treated mice. Eleven control mice showed congestion in the lungs, while none was detected in any of the vepoloxamer-treated animals. All of the vepoloxamer-induced changes were highly significant (see Table 1).
| Pathology | Control | Vepoloxamer treated | Significance* |
| Thrombosis | 13/13 | 13-Feb | < 0.0001 |
| RBC infiltration | 13-Sep | 0/13 | 0.0002 |
| Intervening septal wall damaged | 13-Sep | 0/13 | 0.0002 |
| RBC in alveolar space | 13-Aug | 0/13 | 0.0008 |
| Congestion | 13-Nov | 0/13 | < 0.0001 |
Table 1: Effect of Vepoloxamer On Lung Pathology in Experimental ACS.
*two-tailed p-value using Fisher’s exact test
The results demonstrate that vepoloxamer decreases mortality and prevents the development of ACS-like pulmonary pathology in a relevant model of acute sickling induced ACS.
CLINICAL EXPERIENCE WITH VEPOLOXAMER IN ACS
The safety of vepoloxamer was studied as part of an open label dose escalation trial in 43 SCD patients diagnosed with ACS [48]. The diagnosis of ACS required that a subject had confirmed SCD, a sudden onset of acute chest pain or respiratory distress lasting at least 4 hours, and a new clinically significant infiltrate on chest X-ray. Eligible subjects received total doses of vepoloxamer between 1,120-2,960mg administered during a 24 hour window by continuous intravenous infusion. Five dose levels were studied with a minimum of six patients at each dose level. Safety was assessed by clinical outcomes and clinical laboratory measures including special renal function tests.
Overall vepoloxamer was well tolerated at all dose levels. The study paid particular attention to renal function since renal dysfunction had been observed in earlier studies investigating unpurified poloxamer in myocardial infarction patients. In marked contrast to those earlier studies, no clinically significant dose-related increase in serum creatinine were observed. In addition, there were no dose-related or clinically relevant changes in specialized biomarkers of renal function (i.e., glomerular integrity, permeability and filtration; proximal tubular injury; distal tubular transport, and protein absorption). As had been reported in previous studies of vepoloxamer in sickle cell patients, [49,50] liver function tests, especially bilirubin and serum transaminases, were mild to moderately elevated during vepoloxamer infusion, but, resolved to baseline during follow-up (usually by day 5-10 but no later than day 28-35 post-infusion). The authors concluded that the administration of vepoloxamer to patients with ACS was safe.
Although the study was not designed to evaluate efficacy, trends in biomarkers and clinical outcomes suggestive of benefit were observed. The degree of sPLA2 is reported to correlate with measures of clinical severity in ACS and the rise in sPLA2 concentrations coincides with the onset of ACS [51]. For all dosages, all patients with elevated sPLA2 showed a significant decrease by the end of vepoloxamer infusion which also continued to decrease during the remainder of the study. Both adult and pediatric patients showed similar trends in sPLA2 concentrations [52]. In addition, there were trends, particularly at higher doses and in children, towards a shorter duration of an ACS episode and the length of ACS hospitalization especially compared to historical controls.
DISCUSSION AND CONCLUSION
Acute Chest Syndrome is one of the most frequent complications in SCD, and together with pulmonary hypertension, is the disease’s most common cause of death. Despite its clinical impact, treatment alternatives for ACS are limited, and there is an unmet need for new prophylactic or therapeutic interventions. In this document, we provide justification for use of vepoloxamer in ACS. Our most compelling argument is based on two underlying and complimentary considerations: First, because multiple pathophysiological processes underlie ACS, it may be necessary to address several processes for optimal outcomes. Vepoloxamer may uniquely achieve this. Second, vepoloxamer appears safe and well tolerated in ACS patients at exposures equaling or exceeding those that result in all the desired pharmacological activity. These and other considerations are discussed more completely below.
In this document we present data showing that vepoloxamer has the capacity to improve many of the biological processing which appear to account for ACS pathophysiology. We presented data showing it improves the deformability and filterability of sRBC thus improving their capacity to move through narrow channels akin to what these cells encounter in vivo within the SCD microvasculature. We also discussed vepoloxamer’s effect in reducing whole blood viscosity under low shear rates. Other published reports confirm this activity [26,53]. The benefits of this rheological activity on blood flow have been demonstrated in vivo during clinical VOC. Administration of vepoloxamer to SCD patients during VOC, significantly improved RBC velocity by 2 hours following start of infusion in all vepoloxamer treated subjects, but in none of the controls. Additional improvement were also observed 7 hours post-infusion, such that the RBC velocity had returned to values similar to those observed at steady state in all vepoloxamer treated subjects but in none of the controls [54]. As mentioned previously, pulmonary vaso-occlusion and reduced blood flow is recognized as the common pathophysiological event initiating the clinical manifestation of ACS. Whether this is driven primarily through enhanced cellular adhesion [55] or by elevated RBC aggregation is a matter of contention [56]. Data presented in this publication that vepoloxamer reduces both of these underlying pathologies.
We also discussed the role of hemolysis in ACS and presented results showing that vepoloxamer stabilizes RBC membranes from hemolysis. This data is consistent with an extensive body of literature showing that vepoloxamer and its unpurified parent poloxamer 188 protects the membranes of a wide variety of cells from mechanical and chemical injury [12,26]. This suggests that vepoloxamer may not only protect injured sRBC from hemolysis, but also protect endothelium and other tissues from ischemic and reperfusion injury as suggested by Hunter [26].
The ability to protect cells from mechanical trauma may also contribute to vepoloxamer’s reported anti-thrombotic activity. By stabilizing RBC from shear-induced membrane damage, vepoloxamer may prevent ADP leakage, and thus ADP induced platelet aggregation / thrombosis. This mechanism may also account for the reported activity in antagonizing the procoagulant actions of microparticles.
Some of the most compelling evidence as to the potential utility of vepoloxamer in treating ACS comes from the studies conducted in human HbS transgenic animals. Of particular interest were the pathology results demonstrating the marked reduction in cellular infiltration and thrombosis with vepoloxamer treatment. These effects are consistent with vepoloxamer’s antithrombotic effects in other experimental models. The role of thrombosis in ACS remains a subject of speculation. However, thrombosis of pulmonary vessels is a common finding on autopsy of patients with SCD and pulmonary artery thrombosis was observed at a prevalence of 17% in a recent analysis of ACS patients [57]. These findings suggest that antithrombotic therapy with an agent like vepoloxamer may be an important therapeutic goal in ACS.
Our document concludes with a discussion of a phase I safety study conducted with vepoloxamer in sickle cell patients experiencing ACS [48]. The study investigated levels of exposure within the range of concentrations demonstrating the pharmacological activities discussed in this publication. At all levels of drug exposure, vepoloxamer was well tolerated and appeared safe. While the study was not designed to evaluate efficacy, trends toward reduced hospitalization and reductions in sPLA2 biomarkers were observed. The safety observed in this study is further supported by an extensive battery of non-clinical toxicology studies on vepoloxamer. Included were studies where vepoloxamer was administered to rats and dogs for 28 consecutive days as a continuous intravenous infusion. In those studies the No Observable Adverse Effect Level (NOAEL) for total exposure was 20,160mg/kg in dogs and 10,080mg/kg in rats. Considering that the anticipated total acute exposure of velopoxamer for treatment of ACS is in the range of 1,500-2,500mg/kg, we predict a several fold safety factor with regard to exposure.
The results from the Phase III EPIC study with vepoloxamer are anticipated to provide further justification for evaluating its use in treating ACS.
CONCLUSION
In ACS (and other sickle related vas-oocclusive events) there are multiple pathological processes including dysfunctional coagulation, inflammation, inappropriate cellular adhesion, hemolysis and others that contribute to ischemia and tissue injury. A common denominator to these seemingly disparate pathways may be the exposure of normally hidden hydrophobic phospholipids in damaged cell membranes serving as activation surfaces for the aforementioned pathological cascades. An agent such as vepoloxamer that will bind to such surfaces instantly sealing the damaged membrane and restoring normal surface hydration characteristics may thus inhibit multiple pathological pathways via a single mechanistic activity. The lack of such damaged surfaces in healthy tissues may also account for vepoloxamers safety profile and the observations that pharmacologically active concentrations of vepoloxamer appear safe and well tolerated in SCD patients with ACS.
This new type of pharmacological strategy holds significant promise for the treatment of ACS. Additional clinical studies to demonstrate its safety and efficacy are warranted.
REFERENCES
- Bunn HF (1997) Pathogenesis and treatment of sickle cell disease. N Engl J Med 337: 762-769.
- Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, et al. (1997) Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative study of sickle cell disease. Blood 89: 1787-1792.
- Castro O, Brambilla DJ, Thorington B, Reindorf CA, Scott RB, et al. (1994) The acute chest syndrome in sickle cell disease: incidence and risk factors. The cooperative study of sickle cell disease. Blood 84: 643-649.
- Parhar K, Parizkova B, Jones N, Valchanov K, Fowles J, et al. (2015) Extracorporeal membrane oxygenation for the treatment of adult sickle cell acute chest syndrome. Perfusion.
- Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, et al. (2000) Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. N Engl J Med 342: 1855-1865.
- Paul RN, Castro OL, Aggarwal A, Oneal PA (2011) Acute chest syndrome: sickle cell disease. Eur J Haematol 87: 191-207.
- Gladwin MT, Vichinsky E (2008) Pulmonary complications of sickle cell disease. N Engl J Med 359: 2254-2265.
- Machado RF, Mack AK, Martyr S, Barnett C, Macarthur P, et al. (2007) Severity of pulmonary hypertension during vaso-occlusive pain crisis and exercise in patients with sickle cell disease. Br J Haematol 136: 319-325.
- Ghosh S, Adisa OA, Chappa P, Tan F, Jackson KA, et al. (2013) Extracellular hemin crisis triggers acute chest syndrome in sickle mice. J Clin Invest 123: 4809-4820.
- Emanuele M, Balasubramaniam B (2014) Differential effects of commercial-grade and purified poloxamer 188 on renal function. Drugs R D 14: 73-83.
- Hunter RL, Luo AZ, Zhang R, Kozar RA, Moore FA (2010) Poloxamer 188 inhibition of ischemia/reperfusion injury: evidence for a novel anti-adhesive mechanism. Ann Clin Lab Sci 40: 115-125.
- Maskarinec SA, Wu G, Lee KY (2005) Membrane sealing by polymers. Ann N Y Acad Sci 1066: 310-320.
- Sandez-Macho I, Casas M, Lage EV, Rial-Hermida MI, Concheiro A, et al. (2015) Interaction of poloxamine block copolymers with lipid membranes: Role of copolymer structure and membrane cholesterol content. Colloids Surf B Biointerfaces 133: 270-277.
- Cheng CY, Wang JY, Kausik R, Lee KY, Han S (2012) Nature of interactions between PEO-PPO-PEO triblock copolymers and lipid membranes: (II) role of hydration dynamics revealed by dynamic nuclear polarization. Biomacromolecules 13: 2624-2633.
- Wu G, Majewski J, Ege C, Kjaer K, Weygand M, et al. (2005) Interaction between lipid monolayers and poloxamer 188: An X-ray reflectivity and diffraction study. Biophys J 89: 3159-3173.
- Burgess SE, Zhao Y, Sen A, Hui SW (2007) Resealing of electroporation of porcine epidermis using phospholipids and poloxamers. Int J Pharm 336: 269-275.
- Frey SL, Zhang D, Carignano MA, Szleifer I, Lee KY (2007) Effects of block copolymer’s architecture on its association with lipid membranes: experiments and simulations. J Chem Phys 127: 114904.
- Ham TH, Castle W (1940) Relation of increased hypotonic fragility and of erythrostasis to the mechanism of hemolysis in certain anemias. Transactions of the Association of American Physicians 55: 127-132.
- Chien S, Usami S, Bertles JF (1970) Abnormal rheology of oxygenated blood in sickle cell anemia. J Clin Invest 49: 623-634.
- Charache S, de la Monte S, MacDonald V (1982) Increased blood viscosity in a patient with sickle cell anemia. Blood Cells 8: 103-109.
- Horne MK 3rd (1981) Sickle cell anemia as a rheologic disease. Am J Med 70: 288-298.
- Tripette J, Alexv T, Hardy-Dessources M, Mougenel D, Beltan E, et al. (2009) Red blood cell aggregation, aggregate strength and oxygen transport potential of blood are abnormal in both homozygous sickle cell anemia and sickle-hemoglobin C disease. Haematologica 94: 1060-1065.
- Connes P, Alexy T, Detterich J, Romana M, Hardy-Dessources MD, et al. (2016) The role of blood rheology in sickle cell disease. Blood Rev 30: 111-118.
- Noguchi CT, Torchia DA, Schechter AN (1982) Determination of sickle hemoglobin polymer in SS and AS erythrocytes. Blood Cells 8: 225-235.
- Hiruma H, Noguchi CT, Uyesaka N, Hasegawa S, Blanchette-Mackie EJ, et al. (1995) Sickle cell rheology is determined by polymer fraction--not cell morphology. Am J Hematol 48: 19-28.
- Hunter RL (2010) Unpublished monograph (poloxamer 188 and purified poloxamer 188).
- Meiselman HJ, Johnson CS (1991) Unpublished Report.
- Gustafsson L, Appelgren L, Myrvold HE (1981) Blood flow and in vivo apparent viscosity in working and non-working skeletal muscle of the dog after high and low molecular weight dextran. Circ Res 48: 465-469.
- Richardson SG, Breeze GR, Stuart J (1976) Hyperfibrinogenaemia and hyperviscosity in sickle-cell crisis. J Clin Pathol 29: 890-893.
- Lamarre Y, Romana M, Waltz X, Lalanne-Mistrih ML, Tressières B, et al. (2012) Hemorheological risk factors of acute chest syndrome and painful vaso-occlusive crisis in children with sickle cell disease. Haematologica 97: 1641-1647.
- Styles LA, Schalkwijk CG, Aarsman AJ, Vichinsky EP, Lubin BH, et al. (1996) Phospholipase A2 levels in acute chest syndrome of sickle cell disease. Blood 87: 2573-2578.
- Srair HA, Owa JA, Aman HA, Madan MA (1995) Acute chest syndrome in children with sickle cell disease. Indian J Pediatr 62: 201-205.
- Baskurt OK, Meiselman HJ (2008) RBC aggregation: more important than RBC adhesion to endothelial cells as a determinant of in vivo blood flow in health and disease. Microcirculation 15: 585-590.
- Hebbel RP, Boogaerts MA, Eaton JW, Steinberg MH (1980) Erythrocyte adherence to endothelium in sickle-cell anemia. A possible determinant of disease severity. N Engl J Med 302: 992-995.
- Telen MJ (2007) Role of adhesion molecules and vascular endothelium in the pathogenesis of sickle cell disease. Hematology Am Soc Hematol Educ Program .
- Stuart MJ, Setty BN (1999) Sickle cell acute chest syndrome: pathogenesis and rationale for treatment. Blood 94: 1555-1560.
- Hsu L, Liu X, Pierangeli S, Eckman J, Aguayo S, et al. (2000) Microcirculatory effects of blocking cell adhesion molecules in transgenic sickle mice. Blood 96: 528a.
- Hines PC, White J, Gao X, Liu K, Emanuele M (2015) Evaluation of purified poloxamer (vepoloxamer) on sickle Red Blood Cell (RBC) adhesion and membrane fragility utilizing microfluidic pulsatile flow adhesion bioassays. 9th Annual Foundation for Sickle Cell Disease Research & Education Symposium and 38th National Sickle Cell Disease Scientific Meeting April, Mast therapeutics, San Diego, CA, USA.
- Taylor JG 6th, Nolan VG, Mendelsohn L, Kato GJ, Gladwin MT, et al. (2008) Chronic hyper-hemolysis in sickle cell anemia: association of vascular complications and mortality with less frequent vasoocclusive pain. PLoS One 3: 2095.
- Kato GJ, Gladwin MT, Steinberg MH (2007) Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev 21: 37-47.
- Baek E, Kim HS, Kim JH, Kim N, Kim H (2009) Stroma-free mass production of clinical-grade Red Blood Cells (RBCs) by using poloxamer 188 as an RBC survival enhancer. Transfusion 49: 2285-2295.
- Adams-Graves P, Kedar A, Koshy M, Steinberg M, Veith R, et al. (1997) RheothRx (poloxamer 188) injection for the acute painful episode of sickle cell disease: a pilot study. Blood 90: 2041-2046.
- Ataga KI, Orringer EP (2003) Hypercoagulability in sickle cell disease: a curious paradox. Am J Med 115: 721-728.
- van Beers EJ, Schaap MC, Berckmans RJ, Nieuwland R, Sturk A, et al. (2009) Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica 94: 1513-1519.
- Armstrong JK, Meiselman HJ, Fisher TC (1995) Inhibition of red blood cell-induced platelet aggregation in whole blood by a nonionic surfactant, poloxamer 188 (RheothRx injection). Thromb Res 79: 437-450.
- Fareed J, Hoppensteadt D, Emanuele M, Syed D, Schuharazad A, et al. (2014) Procoagulant Actions of Circulating Microparticles in Sickle Cell Anemia and Sepsis Associated Coagulopathy and Their Modulation by a Triblock Polymer MST 188. Blood
- Robinson K, Hunter R, Stack J, Hearn J, Apkarian R, Roubin G (1990) Inhibition of Coronary Arterial Thrombosis in Swine By Infusion of Poloxamer 188 J Inv Car 2: 9-20.
- Ballas SK, Files B, Luchtman-Jones L, Benjamin L, Swerdlow P, et al. (2004) Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin 28: 85-102.
- Orringer EP, Casella JF, Ataga KI, Koshy M, Adams-Graves P, et al. (2001) Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease: A randomized controlled trial. JAMA 286: 2099-2106.
- Adams-Graves P, Kedar A, Koshy M, Steinberg M, Veith R, et al. (1997) RheothRx (poloxamer 188) injection for the acute painful episode of sickle cell disease: a pilot study. Blood 90: 2041-2046.
- Styles LA, Aarsman AJ, Vichinsky EP, Kuypers FA (2000) Secretory phospholipase A(2) predicts impending acute chest syndrome in sickle cell disease. Blood 96: 3276-3278.
- Ballas SK, Files B, Luchtman-Jones L, Benjamin L, Swerdlow P, et al. (2006) Secretory phospholipase A2 levels in patients with sickle cell disease and acute chest syndrome. Hemoglobin 30: 165-170.
- Carter C, Fisher TC, Hamai H, Johnson CS, Meiselman HJ, et al. (1992) Haemorheological Effects of a Nonionic Copolymer Surfactant (Poloxamer 188). Clinical Hemorheology and Microcirculation 12: 109-120.
- Cheung A, Chan MS, Ramanujam S, Rangaswami A, Curl K, et al. (2004) Effects of poloxamer 188 treatment on sickle cell vaso-occlusive crisis: computer-assisted intravital microscopy study. J Investig Med 52: 402-406.
- Yedgar S, Kaul DK, Barshtein G (2008) RBC adhesion to vascular endothelial cells: more potent than RBC aggregation in inducing circulatory disorders. Microcirculation 15: 581-583.
- Baskurt OK, Meiselman HJ (2008) RBC aggregation: more important than RBC adhesion to endothelial cells as a determinant of in vivo blood flow in health and disease. Microcirculation 15: 585-590.
- Mekontso Dessap A, Deux JF, Abidi N, Lavenu-Bombled C, Melica G, et al. (2011) Pulmonary artery thrombosis during acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med 184: 1022-1029.
Citation: Emanuele M, McKenzie D (2016) The Pharmacologic Rationale Supporting the Potential Use of Vepoloxamer for the Treatment of Acute Chest Syndrome in Patients with Sickle Cell Disease. J Hematol Blood Transfus Disord 3: 006
Copyright: © 2016 Martin Emanuele, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.