Journal of Ophthalmology & Clinical Research Category: Clinical
Type: Case Report
An Exophthalmic Clinical Presentation of Cementomatous Fibrous Dysplasia in an Orbital Roof
*Corresponding Author(s):
Myron YanoffOphthalmology Department, School Of Medicine, University Of Pennsylvania, Drexel University College Of Medicine, Philadelphia, United States
Tel:+1 2157623937,
Email:myron.yanoff@drexelmed.edu
Received Date: Apr 05, 2017
Accepted Date: May 12, 2017
Published Date: May 26, 2017
Abstract
A 10-year-old boy with no prior ocular complaints presented to the St. Christopher’s Hospital for Children in Philadelphia with slowly progressing right eye proptosis. Computed tomography and magnetic resonance imaging of the head and brain showed a right orbital roof intraosseous mass displacing the superior rectus muscle, causing right exophthalmos. Debulking of the lesion was performed. Pathology diagnosed cementomatous variant of fibrous dysplasia with a genetic component of loss of chromosomal segments at XQ and 2Q, specifically 2q33.1 and Xq26.3q27.1. Chromosomal losses at Xq26 and 2q33 were described in three cases of cemento-ossifying fibromas by Sawyer et al., in 1995. We provide supporting evidence for a possible cytogenetic relationship between Xq26 and 2q33 chromosomal losses and the occurrence of fibro-osseous lesions. Based on the patient’s history, ocular and histological examination, exophthalmos occurred in this patient due to orbital roof cementomatous variant of fibrous dysplasia. To the best of our knowledge, there have been no previously reported cases or cytogenetic findings associated with a cementomatous variant of fibrous dysplasia in the orbital roof.
Keywords
Cementomatous variant; Exophthalmos; Fibrous dysplasia
INTRODUCTION
Fibrous Dysplasia (FD) is depicted as a benign, slow growing fibro-osseous lesion presenting in skeletal bone that can be monostotic or polyostotic in nature [1-3]. When presenting in the craniomaxillofacial region, specifically the orbit, it can cause painless bony enlargement producing ocular manifestations of exophthalmos, deformity, and visual problems. Neuroimaging is essential to identify the extent of bony involvement. Fibrous dysplasia should be high on the differential for a pediatric ophthalmologist as it typically presents in the first 3 decades of life. Malignant transformation is unlikely in children, but FD can cause visual loss especially when involving compression of the optic nerve. There have been no previously reported findings of a cytogenetic or genetic mutation associated with monostotic fibrous dysplasia lesions.
CLINICAL PRESENTATION AND DIAGNOSTIC IMAGING
A 10-year-old boy presented to the St. Christopher’s Hospital for Children Ophthalmology Department in Philadelphia, PA in October 2015 as a new patient. The chief complaint was a protruding right eye noted by the patient and his parents. The right exophthalmos had been occurring over a 3-month period. Upon arrival to the ophthalmology department, the patient was immediately sent to the Neurosurgery department with notification that the patient had slowly progressing proptosis of his right eye in need of Neuroimaging (Figures 1 and 2).
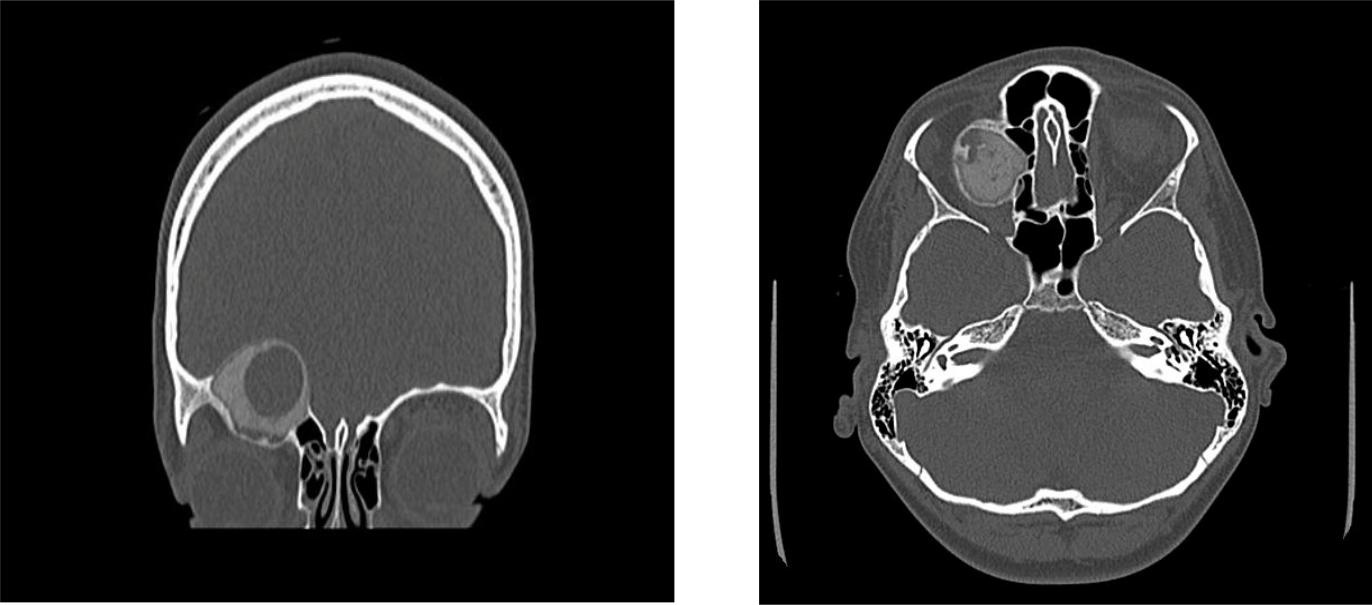
Figure 1A & 1B: Preoperative CT scan of the orbits showing a well-circumscribed, ground glass, calcific, osseous, expansile mass with a cystic component measuring, 2.7 x 3.0 x 4.0, centered within the right orbital roof.
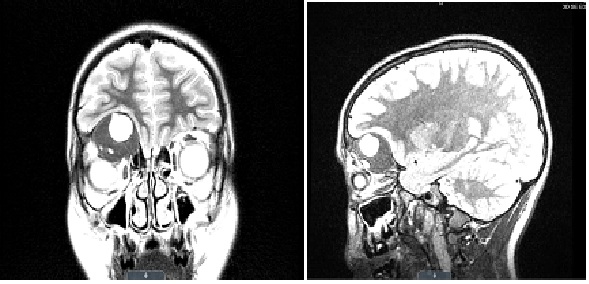
Figure 2A & 2B: MRI described the mass with similar measurements to have a 1.5 cm multi-septated, cystic-fluid filled, necrotic center with mass effect on the inferior frontal lobe while flattening and inferiorly displacing the superior rectus muscle of the right globe, causing the noted proptosis. The right globe and optic nerve were intact in both CT and MRI. Differential diagnosis based on imaging was fibrous dysplasia, rhabdomyosarcoma, Langerhans cell histocytosis and Lymphoma/Leukemia. The patient was immediately scheduled for a right frontal craniotomy/orbitotomy for intraosseous tumor debulking procedure performed by neurosurgery and plastic surgery.
Based on the operative report, the tumor was visible after removal of the orbital rim [4]. The tumor was internally debulked confirming its intraosseous origin comprised of bone and soft tissue and sent for frozen and permanent sections [4]. Debulking continued circumferentially until mass effect was released from the frontal lobe and orbit [4]. The frontal sinus was exonerated by removing mucosa [4]. The remaining orbital rim was thoroughly scraped with a curette to remove any additional tumor contents [4] (Figure 3).
Based on the operative report, the tumor was visible after removal of the orbital rim [4]. The tumor was internally debulked confirming its intraosseous origin comprised of bone and soft tissue and sent for frozen and permanent sections [4]. Debulking continued circumferentially until mass effect was released from the frontal lobe and orbit [4]. The frontal sinus was exonerated by removing mucosa [4]. The remaining orbital rim was thoroughly scraped with a curette to remove any additional tumor contents [4] (Figure 3).
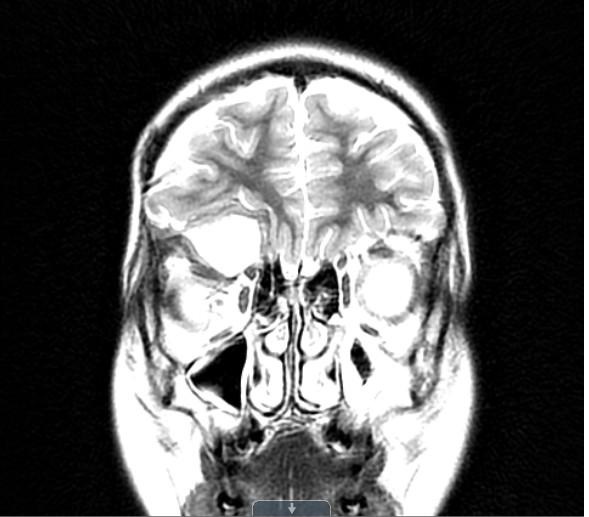
Figure 3: MRI imaging after craniotomy/orbitotomy: January 2016.
A residual, expansile, heterogeneous mass centered in the roof of the right orbit with an internal cystic component and strong heterogeneous peripheral enhancement, measuring approximately 3.4 cm AP x 3.4 cm TR and 2.4 cm CC, causing mild right proptosis and mass effect on the right frontal lobe while in direct contact with the superior rectus muscle.
Multiple attempts were made to call the patient with no response. Patient was lost to follow up after his visit in January 2016.
HISTOLOGY
The Pathology department at St. Christopher’s Hospital for Children received specimens measuring 1 x 0.8 by 0.3 cm and 3 x 2.5 x 1.8 cm. Histological examination of the lesion was performed by Dr Matthew Keisling with consultation to Dr Edward McCarthy at Johns Hopkins Reference Laboratories. The diagnosis was fibro-osseus lesion consistent with fibrous dysplasia, cementomatous variant, also referred to as cementomatous fibrous dysplasia (Figures 4a-4d).
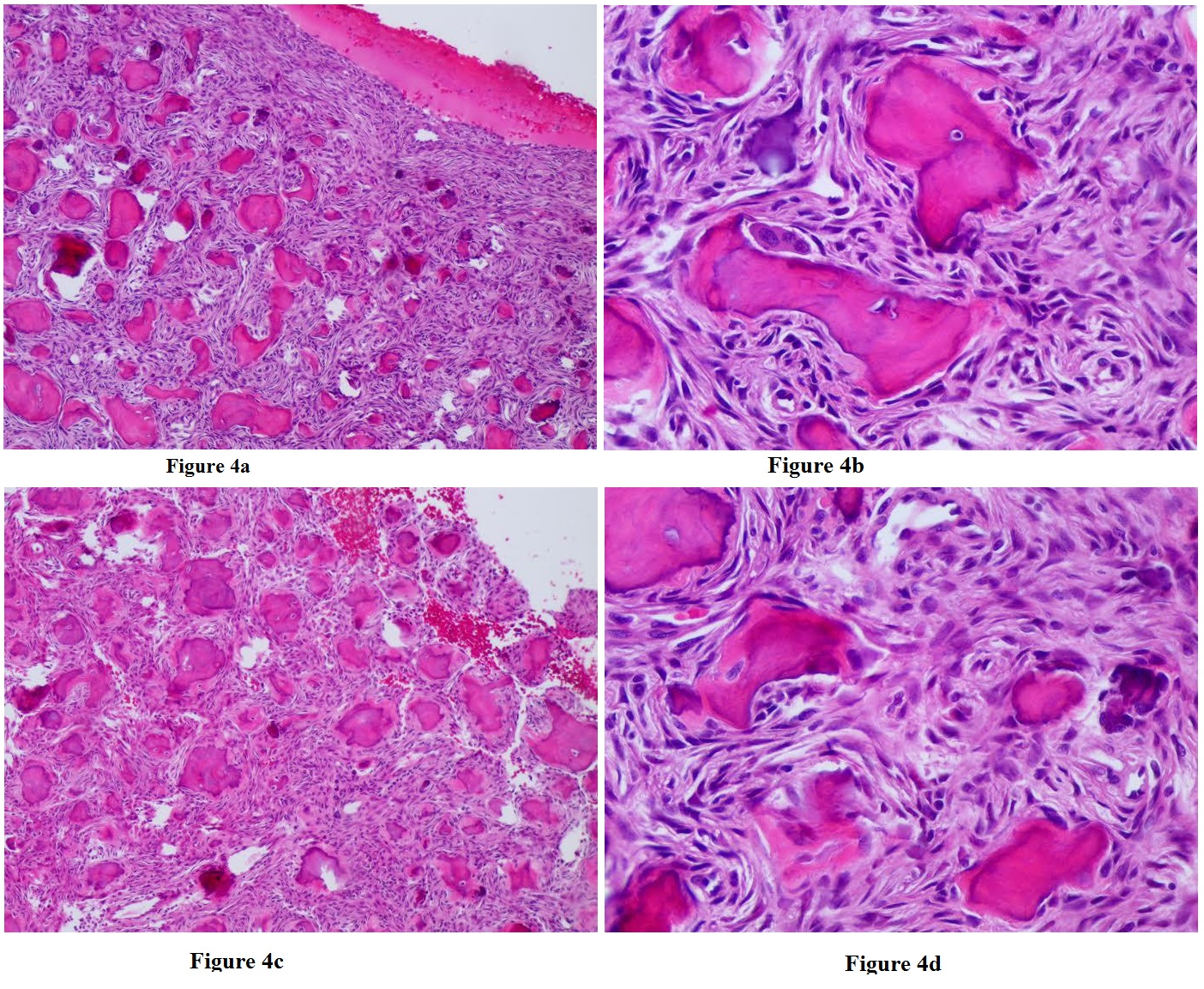
Figure 4a-4d: A low power view of immature woven bone composed of irregularly curved trabeculae surrounded by fibroblastic stroma are shown in 4a and 4b, with higher magnification shown in 4c & 4d (H & E stain) Histology Courtesy of Matthew Keisling, DO.
GENETICS
Cytogenetics was performed on our patient and he was noted to have loss of chromosomal segments at XQ and 2Q, more specifically 2q33.1 (199,955,317-200,424,710) x1, Xq21.31 (89,752,139-91,890,838) x0, Xq26.3q27.1 (137,096,830-139,059,830) x0, and Xq27.1q27.3 (139,791,912-144,559,812) x0. Fibro-osseous lesions include fibrous dysplasia, ossifying fibroma, cemento-ossifying fibroma and osteo-fibrous dysplasia. The cause of fibrous dysplasia is not well understood, however these cytogenetic findings suggest there is a genetic association linking monostotic fibro-osseous lesions. The same chromosomal losses in Xq26 and 2q33 found in our patient were characteristically described in only one other article published by Sawyer et al., [5]. In Cancer 1995, Sawyer et al., identified three cases of cemento-ossifying fibromas in the orbit wall with non-random breakpoints at bands Xq26 and 2q33. Sawyer highlighted the clinical importance of finding consistent structural chromosomal abnormalities, since there were only nine cytogenetic cases of histologically benign fibro-osseous lesions reported prior to 1995. Sawyer et al., highlights that the role of chromosomal aberrations in benign, non-malignant tumors is to identify similar genomic instability to aid in diagnosis and familiarize genetic components that are involved in non-malignant tumor growth [5]. Sawyer et al., described that because there were only three cases identified, it is possible that these chromosomal aberrations occurred by chance; however our case lends support to Sawyer’s cases in recognition that these chromosomal losses at Xq26 and 2q33 are in fact non-random and involved in the tumor growth of fibro-osseous lesions.
DISCUSSION
Fibrous Dysplasia (FD) is depicted as a benign, slow growing fibro-osseous lesion of unknown origin for sporadic cases [6]. FD was first described by von Recklinghausen in 1891 in McCune-Albright Syndrome (MAS) with genetically linked with the GNAS1 gene somatic mutation, however in 1938 Lichtenstein coined the term fibrous dysplasia in its monostotic and polyostotic character [1-3]. FD comprises 2.5% of all bony tumors and 7.5% of benign bony lesions [7]. FD typically involves the skull, long bones, and ribs in which there is replacement of normal bone marrow with fibro-osseous tissue manifesting as a painless asymmetric bony enlargement [1,3,6]. FD is monostotic in 70-80% of cases, while 20-30% are polyostotic, and only 10% of overall cases are found to be in the craniofacial region, most commonly in the maxilla and frontal bone [1,2,6]. The monostotic variant usually presents in the craniofacial bones, femur, tibia and ribs, while the polyostotic form typically presents in the femur, tibia and pelvis with common complaints of pain associated with recurrent fractures [8]. Age predilection is poorly specified in the literature; however FD can typically present in the first 3 decades of life and occurs equally in both sexes [7,9]. When affecting children, fibrous dysplasia often occurs in children older than 2 years of age and can affect adolescents and young adults with 75% of tumors diagnosed before age 30 [6,8]. Findings support more of a dysplastic process rather than neoplastic process, however there has been notation of a 0.4% change of malignant transformation as opposed to the 0.001% chance in normal bone [3]. Malignant transformation was evidenced in Lebeau et al., fibrous dysplasia case of transformation to sarcoma [10]. Nevertheless, Cottalorda et al., argues in a case series of 10 boys and 9 girls with fibrous dysplasia at an average age of 9 years old, none of the cases progressed to malignant transformation [11].
FD is a representation of elements of immature mesenchymal cells (osteoblasts) originating from mutated pluripotent embryonic cells failing to undergo differentiation and maturation [6,12]. Maurie et al., compared osteoblastic activity associated with increased Gsα mutation expression in 2 McCune-Albright Syndrome patients to a patient with a single monostotic FD lesion [13]. Maurie et al., data showed that 30% higher intracellular cAMP, increased proliferation of osteoblastic cells, and lower osteocalcin production were all associated with the Gsα mutation in both monostotic and MAS dysplastic FD cells determining severity of the lesion [13]. Upon gross examination, the lesion may appear gray or bluish gray, and have hemorrhagic or cystic regions composed of yellow serous fluid [8]. Histologically, fibrous dysplasia typically encompasses an osseous component of immature woven bone composed of thin, irregularly shaped trabeculae termed as “alphabet soup” with absent osteoblastic rimming, while the fibrous component surrounds the osseous component with combination of collagen, myxoid, hemorrhagic and cystic components [6,8]. As was exemplified in our patient, there can be a cementifying variant, in which the mineralization is densely composed of concentric, laminated “cementoid” bodies providing the diagnosis of fibrous cementoma or cementomatous variant of fibrous dysplasia [8].
Radiographically, lesions can appear sclerotic in 35% of cases, mixed type in 40% of cases, and least commonly lytic [7]. Computed Tomography (CT) is determined to be the most useful in evaluating craniofacial lesions [1]. CT is the best imaging modality for idenitification of FD due to its bony origin, however Magnetic Resonance Imaging (MRI) is ideal for evaluation of orbital involvement [7]. FD exhibits a “ground glass” appearance due to the woven bone and fibrous component with areas of sclerosis, centered primarily in the medullary canal with cortical expansion [6-8]. Occasionally, there can be areas of cartilaginous differentiation with ring or punctate calcifications, and if prominent, the diagnosis should be fibrocartilaginous dysplasia [8].
Fibrous dysplasia management includes observation if symptoms are minimal to assess for tumor growth, conservative debulking, or radical bony excision and reconstruction [2]. Goisis et al., states that non-surgical approaches results in poor outcomes and surgical treatment is determined by substantial functional decline [3]. Ricalde et al., emphasizes that observation is dependent upon location of the lesion(s), age of the patient, serial clinical examinations and patient’s opinion about surgery [7]. Conservative debulking involves bone contouring with partial resection of localized areas of dysplasia, while radical excision would be supported in a situation of increased risk of malignant transformation, and optic nerve decompression reserved for lesions impinging the optic nerve [3,7]. Giosis et al., supports complete resection although it can be more debilitating regarding quality of life [3]. According to Chen and Noordhoff’s Table of treatment protocol based on dysplasia location, facial area of Zone 1 involving the orbit necessitates radical excision and reconstruction with autogenous bone graft [14]. Moore et al., reviewed sixteen children with orbitocranial fibrous dysplasia that underwent excision of the bony lesion, fifteen of which had craniofacial reconstruction [15]. Orbital wall reconstruction requires precise modeling and alloplastic materials or autologous bone grafts, while orbital roof reconstruction requires separation of orbital contents from the frontal lobe [3]. The focus of reconstruction is to restore contour symmetry, orbital volume, nasolacrimal integrity, and an intact cranial base and barrier [7].
Fibrous dysplasia is often present in children, and furthermore within the craniofacial bones causing ocular symptoms. Leong et al., presented a case report of a 12 year old Asian female with progressive visual loss from a right anterior skull base fibrous dysplasia causing extradural compression of the intracanalicular optic nerve requiring immediate optic nerve decompression with some post-operative visual recovery [9]. Tabrizi et al., described a case of an 8 year old girl whom presented with right side swelling of her face and orbit displacement found to have right maxilla fibrous dysplasia involving the right orbital floor requiring immediate resection and reconstruction [1]. Skorek et al., described an 8 year old boy with inferior orbital wall destruction secondary to fibrous dysplasia [16]. Joseph et al., discussed a case of gradually progressive unilateral exophthalmos and inferolateral displacement of the right globe in a 9 month old infant whom was found to have an extraconal lesion in the superomedial portion of the right orbit with simultaneous destruction of the superomedial orbital wall and roof, histologically confirmed to be fibrous dysplasia [17].
For completion, ossifying fibromas should be included in the differential diagnosis for fibrous dysplasia. Ossifying fibromas are another common fibro-osseous tumor with very different histological components. An ossifying fibroma is characteristically a benign well demarcated fibro-osseous tumor with a sclerotic border typically occurring in the jaw and craniofacial bones with the paranasal sinuses being affected most often [6,18]. When involving the orbit they usually develop from the ethmoid region or superior orbital plate of the frontal bone [6]. Clinical presentation in adults begins in the third to 4th decade of life, while the juvenile type can occur in children and young adults with no sexual predilection [18]. Pathology depicts a well-circumscribed unilobular lesion with low to moderate cellularity with either one or both of two patterned mineralized bone growth: irregular bony trabeculae or psammomatoid pattern. Due to the aggressive growth of ossifying fibroma, it typically requires complete surgical resection, whereas fibrous dysplasia is slow growing and can be self-limited [6]. Wakefield et al., presented the 2nd case of lateral orbital wall presentation of ossifying fibroma, while Sigler et al., presented a case of a 12 year old boy with cementifying fibroma originating in the maxillary, ethmoid, and frontal sinuses resulting in proptosis [19,20]. Fibrous dysplasia is often confused with ossifying fibroma due to similar clinical presentation.
As evidenced, fibrous dysplasia presentation is prominent in the bony orbit with common ocular manifestations in children of various ages ranging from exophthalmos, globe/orbit displacement, facial swelling, headaches/periorbital pain, epiphora and visual disturbance/loss [1,2,6,7,17,]. Literary evidence aptly confirms the importance of being clinically aware of the ocular manifestation of fibrous dysplasia in children of all ages. Early detection of this dysplastic disease process in the pediatric ophthalmologist office can allow for prompt imaging and surgical management of fibrous dysplasia involving the orbit.
FD is a representation of elements of immature mesenchymal cells (osteoblasts) originating from mutated pluripotent embryonic cells failing to undergo differentiation and maturation [6,12]. Maurie et al., compared osteoblastic activity associated with increased Gsα mutation expression in 2 McCune-Albright Syndrome patients to a patient with a single monostotic FD lesion [13]. Maurie et al., data showed that 30% higher intracellular cAMP, increased proliferation of osteoblastic cells, and lower osteocalcin production were all associated with the Gsα mutation in both monostotic and MAS dysplastic FD cells determining severity of the lesion [13]. Upon gross examination, the lesion may appear gray or bluish gray, and have hemorrhagic or cystic regions composed of yellow serous fluid [8]. Histologically, fibrous dysplasia typically encompasses an osseous component of immature woven bone composed of thin, irregularly shaped trabeculae termed as “alphabet soup” with absent osteoblastic rimming, while the fibrous component surrounds the osseous component with combination of collagen, myxoid, hemorrhagic and cystic components [6,8]. As was exemplified in our patient, there can be a cementifying variant, in which the mineralization is densely composed of concentric, laminated “cementoid” bodies providing the diagnosis of fibrous cementoma or cementomatous variant of fibrous dysplasia [8].
Radiographically, lesions can appear sclerotic in 35% of cases, mixed type in 40% of cases, and least commonly lytic [7]. Computed Tomography (CT) is determined to be the most useful in evaluating craniofacial lesions [1]. CT is the best imaging modality for idenitification of FD due to its bony origin, however Magnetic Resonance Imaging (MRI) is ideal for evaluation of orbital involvement [7]. FD exhibits a “ground glass” appearance due to the woven bone and fibrous component with areas of sclerosis, centered primarily in the medullary canal with cortical expansion [6-8]. Occasionally, there can be areas of cartilaginous differentiation with ring or punctate calcifications, and if prominent, the diagnosis should be fibrocartilaginous dysplasia [8].
Fibrous dysplasia management includes observation if symptoms are minimal to assess for tumor growth, conservative debulking, or radical bony excision and reconstruction [2]. Goisis et al., states that non-surgical approaches results in poor outcomes and surgical treatment is determined by substantial functional decline [3]. Ricalde et al., emphasizes that observation is dependent upon location of the lesion(s), age of the patient, serial clinical examinations and patient’s opinion about surgery [7]. Conservative debulking involves bone contouring with partial resection of localized areas of dysplasia, while radical excision would be supported in a situation of increased risk of malignant transformation, and optic nerve decompression reserved for lesions impinging the optic nerve [3,7]. Giosis et al., supports complete resection although it can be more debilitating regarding quality of life [3]. According to Chen and Noordhoff’s Table of treatment protocol based on dysplasia location, facial area of Zone 1 involving the orbit necessitates radical excision and reconstruction with autogenous bone graft [14]. Moore et al., reviewed sixteen children with orbitocranial fibrous dysplasia that underwent excision of the bony lesion, fifteen of which had craniofacial reconstruction [15]. Orbital wall reconstruction requires precise modeling and alloplastic materials or autologous bone grafts, while orbital roof reconstruction requires separation of orbital contents from the frontal lobe [3]. The focus of reconstruction is to restore contour symmetry, orbital volume, nasolacrimal integrity, and an intact cranial base and barrier [7].
Fibrous dysplasia is often present in children, and furthermore within the craniofacial bones causing ocular symptoms. Leong et al., presented a case report of a 12 year old Asian female with progressive visual loss from a right anterior skull base fibrous dysplasia causing extradural compression of the intracanalicular optic nerve requiring immediate optic nerve decompression with some post-operative visual recovery [9]. Tabrizi et al., described a case of an 8 year old girl whom presented with right side swelling of her face and orbit displacement found to have right maxilla fibrous dysplasia involving the right orbital floor requiring immediate resection and reconstruction [1]. Skorek et al., described an 8 year old boy with inferior orbital wall destruction secondary to fibrous dysplasia [16]. Joseph et al., discussed a case of gradually progressive unilateral exophthalmos and inferolateral displacement of the right globe in a 9 month old infant whom was found to have an extraconal lesion in the superomedial portion of the right orbit with simultaneous destruction of the superomedial orbital wall and roof, histologically confirmed to be fibrous dysplasia [17].
For completion, ossifying fibromas should be included in the differential diagnosis for fibrous dysplasia. Ossifying fibromas are another common fibro-osseous tumor with very different histological components. An ossifying fibroma is characteristically a benign well demarcated fibro-osseous tumor with a sclerotic border typically occurring in the jaw and craniofacial bones with the paranasal sinuses being affected most often [6,18]. When involving the orbit they usually develop from the ethmoid region or superior orbital plate of the frontal bone [6]. Clinical presentation in adults begins in the third to 4th decade of life, while the juvenile type can occur in children and young adults with no sexual predilection [18]. Pathology depicts a well-circumscribed unilobular lesion with low to moderate cellularity with either one or both of two patterned mineralized bone growth: irregular bony trabeculae or psammomatoid pattern. Due to the aggressive growth of ossifying fibroma, it typically requires complete surgical resection, whereas fibrous dysplasia is slow growing and can be self-limited [6]. Wakefield et al., presented the 2nd case of lateral orbital wall presentation of ossifying fibroma, while Sigler et al., presented a case of a 12 year old boy with cementifying fibroma originating in the maxillary, ethmoid, and frontal sinuses resulting in proptosis [19,20]. Fibrous dysplasia is often confused with ossifying fibroma due to similar clinical presentation.
As evidenced, fibrous dysplasia presentation is prominent in the bony orbit with common ocular manifestations in children of various ages ranging from exophthalmos, globe/orbit displacement, facial swelling, headaches/periorbital pain, epiphora and visual disturbance/loss [1,2,6,7,17,]. Literary evidence aptly confirms the importance of being clinically aware of the ocular manifestation of fibrous dysplasia in children of all ages. Early detection of this dysplastic disease process in the pediatric ophthalmologist office can allow for prompt imaging and surgical management of fibrous dysplasia involving the orbit.
CONCLUSION
We discussed a case of a 10-year-old boy who presented to St. Christopher’s Hospital in Philadelphia, PA with several months of right eye exophthalmos, found have a right orbital roof lesion comprised of the cementomatous variant of fibrous dysplasia. We offered clinical, histological, surgical and genetic background of the diagnosis. Our case report strives to provide supporting evidence for a possible genetic association with the development of cementomatous fibrous dysplasia in the orbit. In evaluation of a differential diagnosis, we provided background on the varying differences between ossifying fibroma and fibrous dysplasia, as they are clinically similar. Most importantly, we emphasized the significance of including fibrous dysplasia in the differential diagnosis of unilateral exophthalmos for the practicing pediatric ophthalmologist. To our knowledge there have been no previously reported cases of cementomatous variant of fibrous dysplasia presenting in the orbital roof.
ACKNOWLEDGEMENT
Support provided by St. Christopher’s Hospital for Children Departments of Ophthalmology, Pathology, Plastic Surgery and Neurosurgery.
REFERENCES
- Tabizi R, Ozkan BT (2008) Craniofacial Fibrous Dysplasia of the Orbit. J Craniofac Surg 19: 1532-1537.
- Bibby K, McFadzean R (1994) Fibrous Dysplasia of the Orbit. British Journal of Ophthalmology 78: 266-270.
- Goisis M, Biglioli F, Guareschi M, Frigerio A, Mortini P (2006) Fibrous Dysplasia of the Orbital Region: Current Clinical Perspectives in Ophthalmology and Craniomaxillofacial Surgery. Ophthal Plast Reconstr Surg 22: 383-387.
- Narayan P (2015) Procedure: Right frontal craniotomy and orbitotomy for resection and debulking of intraosseous mass. Operative Report, Department of Neurosugery, St. Christopher’s Hospital for Children, Philadelphia, USA.
- Sawyer JR, Tryka AF, Bell JM, Boop FA (1995) Nonrandom Chromosome Breakpoints at Xq26 and 2q33 Characterize Cemento-Ossifying Fibromas of the Orbit. Cancer 76:1853-1859.
- Chung EM, Murphey MD, Specht CS, Cube R, Smirniotopoulos JG (2008) Pediatric Orbit Tumors and Tumorlike Lesions: Osseous Lesions of the Orbit. RadioGraphics 28: 1193-1214.
- Ricalde P, Horswell BB (2001) Craniofacial Fibrous Dysplasia of the Fronto-Orbital Region: A Case Series and Literature Review. J Oral Maxillofac Surg 59: 157-168.
- Crawford BE, Schmeig JJ (2015) Differential Diagnosis in Surgical Pathology 16: 845.
- Leong LT and Ming BJ (2015) Craniofacial Fibrous Dysplasia Involving the Orbit: A Case Report and Literature Review. Asia Pac J Ophthalmol (Phila) 4: 151-154.
- Lebeau J. Fasano D, Antoine P, Pasquier B, Bachelot P, et al. (1984) Malignant Transformation of fibrous dysplasia of the orbit. Rev Stomatol Chir Maxillofac 85: 403-410.
- Cottalorda J, Haddah H, Bolini G, Jouve JL, Muracciole P, et al. (1993) Fibrous Dysplasia in Children. Pediatrie 48: 818-822.
- Parent AD, Shiflett JM (2011) Skull Tumors and Fibrous Dysplasia. In: Youmans Neurological Surgery. 6: 2136-2143.
- Marie PJ, de Pollak C, Chanson P, Lomri A (1997) Increased proliferation of osteoblastic cells expressing the activating Gs alpha mutation in monostotic and polyostotic fibrous dysplasia. Am J Pathol 150: 1059-1069.
- Chen YR, Noordhoff MS (1990) Treatment of craniomaxillofacial fibrous dysplasia: how early and how extensive? Plast Reconstr Surg 86: 835-842.
- Moore AT, Buncic JR, Munro IR (1985) Fibrous Dysplasia of the orbit in childhood. Ophthalmology 92: 12-20.
- Skorek A, Stankiewicz C, Babinski D, Izycka-Swieszewska E (2006) [Fibrous Dysplasia in 8 year old child. Case Report]. Otolaryngol Pol 60: 75-78.
- Joseph E, Kachhara R, Bhattacharya RN, Radhakrishnan VV, Balachandran K (2000) Fibrous dysplasia of the orbit in an infant. Pediatr Neurosurg 32: 205-208.
- Horvai AE, Link T (2012) Bone and Soft Tissue Pathology. Elsevier Health Sciences, Amsterdam, Netherlands.
- Wakefield MJ, Ross AH, Damato EM, Salvi SM, Baker GR (2010) Review of Lateral Orbital Wall Ossifying Fibroma. Orbit 29: 317-320.
- Sigler SC, Wobig JL, Dierks EJ, Dailey RA, Orr JE (1997) Cementifying fibroma presenting as proptosis. Ophthal Plast Reconstr Surg 13: 277-280.
Citation: Brown A, Spector RT, Yanoff M (2017) An Exophthalmic Clinical Presentation of Cementomatous Fibrous Dysplasia in an Orbital Roof. J Ophthalmic Clin Res 4: 028.
Copyright: © 2017 A’sha Brown, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!