Morphological Changes of Glia in Prion and a Prion-Like Disorder
*Corresponding Author(s):
Toledano ACajal Institute, Spanish Research Council (CSIC), Madrid, Spain
Tel:+0034 976762944,
Email:mmonzon@unizar.es
Abstract
Several neurodegenerative diseases such as Alzheimer’s, Parkinson’s and Huntington’s are considered to be prion-like disorders in that they are all proteinopathies where in aberrant proteins spread throughout the brain during disease progression, and thus they may share molecular basis and mechanisms of propagation. Therefore, studies elucidating mechanisms of prion propagation may be relevant to other neurodegenerative diseases. While substantial progress has been made, the pathogenesis of these neurodegenerative diseases is still largely unknown, and as consequence, to date no truly effective treatments that prevent onset or delay progression of these diseases have been identified. In addition to propagation of misfolded proteins, these diseases all induce a host response that includes activation of astrocytes and microglial cells. However, in our opinion, the glial response in each of these diseases has not been well-defined.
Since a role for glial response in prion disease has been clearly demonstrated in a previous study concerning Scrapie in sheep, a similar approach to analysis of astrocytic gliosis has been taken here for Creutzfeldt-Jakob (CJD) and Alzheimer’s Diseases (AD). Here, morphological analysis of glial cells in cerebella from CJD and AD patients (as the most common prion and prion-like disorders, respectively) was performed.
The results presented in this study support the involvement of glial cells not only in the pathogenesis of CJD, but also of AD. A relationship between intensity and morphology is observed in astroglia from the molecular layer in both pathologies. By contrast, the involvement of microgliosis in AD-affected samples showed a lower relevance from that observed in CJD, since reactive microglia were much more abundant in prion disease.
Further analysis of the role of gliosis in CJD and AD, as well as other neurodegenerative diseases, may well advance knowledge of the mechanisms underlying these diseases and may also provide new targets for therapeutic intervention.
Keywords
Alzheimer’s disease; Creutzfeldt-Jakob disease; Glia; Prion diseases
INTRODUCTION
Current evidence supports the idea that many neurodegenerative diseases share specific features with prion diseases. Molecular mechanisms consisting of aggregation and spreading of misfolded proteins that are characteristic for each of them show similarities and, as a result, they have been included in a prion-like disease group [1]. Stanley Prusiner, the first scientist to identify the unique characteristics of prion disease, has even applied the term ‘prion’ to the specific aberrant proteins in each of these diseases that are the pathological hallmark, including ?-amyloid in Alzheimer’s and ?-synuclein in Parkinson’s disease [2,3]. At microscopic level, all these disorders show histopathological insults such as amyloid deposits, neuronal loss, neurite degeneration and gliosis. The present study is particularly focused on contributing to knowledge concerning the latter-gliosis.
Astroglia were initially thought to simply support neurons. More recent research has demonstrated they are essential for correct functioning of the Central Nervous System (CNS). Astrocytes are a cooperative component with neurons in the physiology, metabolism and homeostasis. They play a critical role in establishment of the Blood Brain Barrier (BBB), embryological CNS development, neurotransmission, and tripartite synapsis [4-9]. The other glial cell type, microglia are the main immune cells in CNS [10,11]. In response to any CNS injury, a glial multicellular response is detectable [12]. Specifically in regards to neurodegenerative disease, an association between reactive astroglia and / or microglia with aberrant protein deposits has been described [13-16]. However, in our opinion, the role of these cellular populations in the neurodegenerative process has not been sufficiently elucidated. Astroglial and microglial subpopulations may have dual roles in the neurodegenerative disorders, exerting both neuroprotective and neurotoxic effects in CNS [6,11,17].
The present study was initiated as a result of our interesting findings concerning involvement of glial cells in neurodegeneration in sheep affected with Scrapie, a natural prion disorder [18,19]. The main aim of the study here was to compare and contrast morphological alterations of glial cells in cerebella from Creutzfeldt-Jakob Disease (CJD, as the most common prion disorder) and Alzheimer’s Disease (AD, as the most common prion-like disorder) as revealed by immunohistochemical labeling with specific markers for astrocytes and microglia.
MATERIAL AND METHODS
Samples
GFAP immunohistochemistry
Morphology and intensity (high, medium, or low) of GFAP immunostaining were assessed in each layer from all the cerebella.
Reactive microglia immunohistochemistry
Morphology and intensity (high, medium, or low) of reactive microglia immunostaining were assessed in each layer from all the cerebella.
RESULTS
GFAP immunostaining
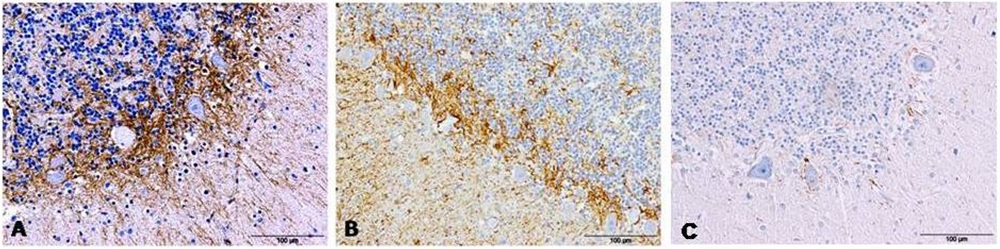 Figure 1: GFAP detection by immunohistochemistry in cerebella from AD (A), CJD (B) and control (C) cases show an evident increase of immunolabeling around Purkinje cells.
Figure 1: GFAP detection by immunohistochemistry in cerebella from AD (A), CJD (B) and control (C) cases show an evident increase of immunolabeling around Purkinje cells.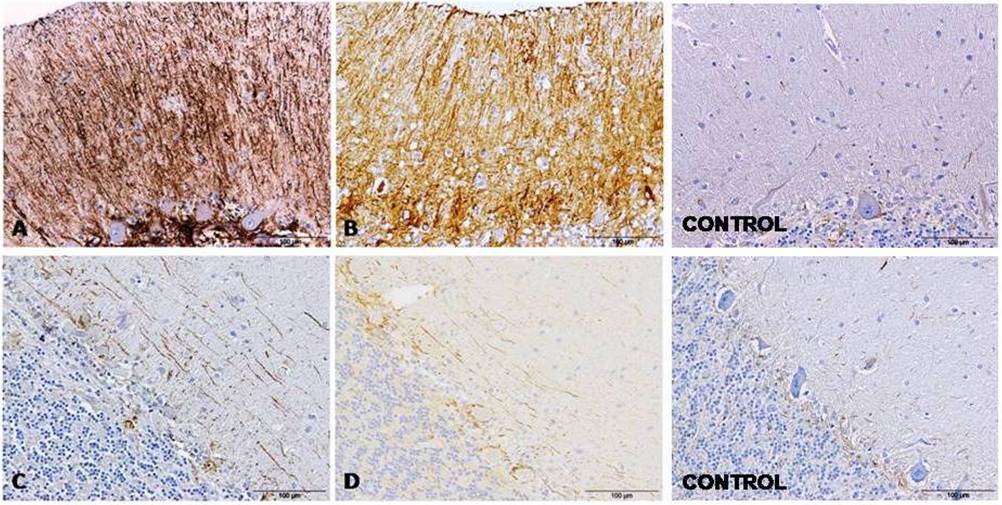 Figure 2: GFAP immunolabeling in cerebella evidenced two different astroglial profiles in the molecular layer from samples affected by both pathologies, AD (A and C) and CJD (B and D), respectively: a radial profile when high GFAP intensity (A and B) or a horizontal profile when lower GFAP intensity were detected (C and D). GFAP immunolabeling in cerebella from control cases are also showed.
Figure 2: GFAP immunolabeling in cerebella evidenced two different astroglial profiles in the molecular layer from samples affected by both pathologies, AD (A and C) and CJD (B and D), respectively: a radial profile when high GFAP intensity (A and B) or a horizontal profile when lower GFAP intensity were detected (C and D). GFAP immunolabeling in cerebella from control cases are also showed.GFAP immunolabeling was consistently detected in association with protein aggregates in both, AD and CJD tissues (Figure 3). Also in both CJD and AD tissue, GFAP immunolabeling appeared next to the meninges in many occasions (Figure 4).
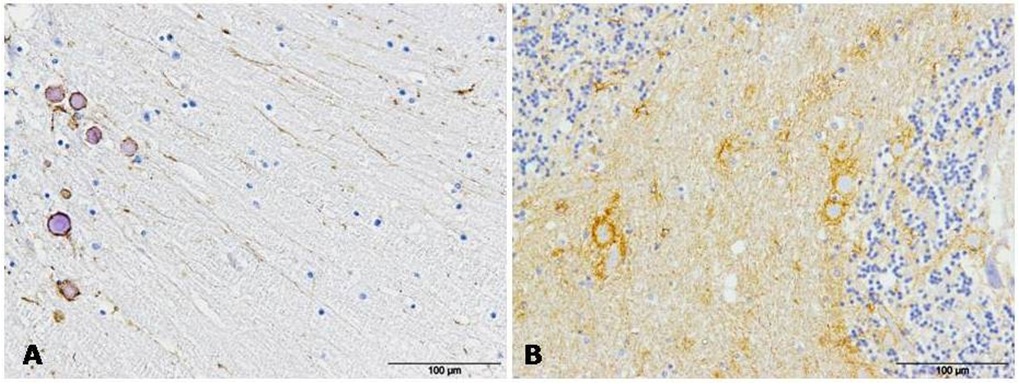 Figure 3: GFAP immunolabeling was always detected in association with protein aggregates in AD (A) and CJD (B) samples.
Figure 3: GFAP immunolabeling was always detected in association with protein aggregates in AD (A) and CJD (B) samples.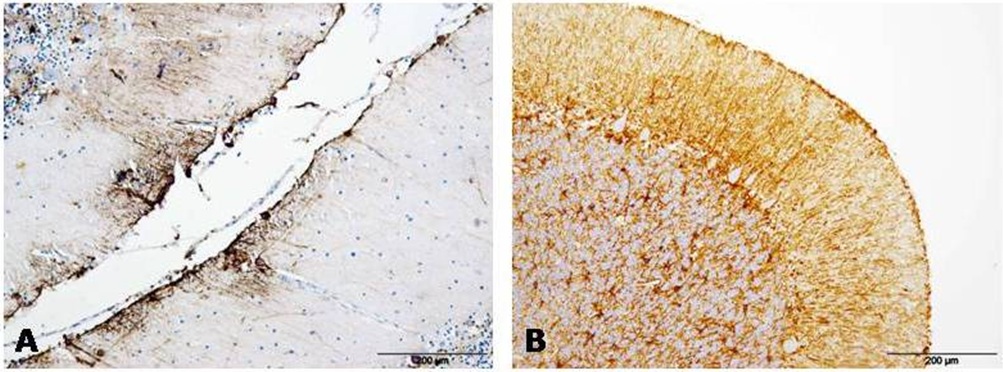 Figure 4: GFAP immunostaining appeared next to meninges in AD (A) and CJD (B) cerebella in many occasions.
Figure 4: GFAP immunostaining appeared next to meninges in AD (A) and CJD (B) cerebella in many occasions.Reactive microglia immunostaining
 Figure 5: Reactive microglia immunostaining in cerebellum differs depending on the pathology studied: scarce, mostly ramified and located in white matter from AD samples (A); or abundant, combined ramified -amoeboid and extended over all the layers from CJD patients (B).
Figure 5: Reactive microglia immunostaining in cerebellum differs depending on the pathology studied: scarce, mostly ramified and located in white matter from AD samples (A); or abundant, combined ramified -amoeboid and extended over all the layers from CJD patients (B).DISCUSSION
Both astrogliosis and microglial activation consistently appear in brain tissue from patients suffering from neurodegenerative disease [6,8,12]. Despite constituting a universal finding across many neurodegenerative diseases, there has not been the focus on this response that is afforded the proteins that are implicated in each disease. The present study constitutes a preliminary approach to determine whether glial changes observed in a human prion disease, CJD, agree with those observed in the most prevalent prion - like disease, AD. To our knowledge, this is the first morphological study performed with the aim to compare the specific alterations of glial cells in these two neurodegenerative diseases. We focused on microglia and astroglia, due to their roles in immune function and therefore, in neuroinflammation. Neuroinflammation itself is considered either causative or contributory to the pathogenesis of neurodegeneration [6,20].
Cerebellum is a brain area affected in all CJD patients, but in AD patient involvement of this area is less evident. However, results presented here indicate that astroglia show similar morphological changes in cerebellum from patients suffering either pathology. Labeling around Purkinje cells had been seen previously in Scrapie-affected sheep [19] and is seen again here in both CJD and AD. In cerebella from AD affected patients, neuronal loss and astrocytosis has previously been assessed, but in that study only familial and sporadic AD were compared [21]. The demonstration here that Purkinje cells are the most ‘protected’ neurons have been evidenced by ultrastructural studies in Scrapie, where they were revealed to be the most damaged neurons too. Vacuolation preferentially occurred around these neurons [22]. This finding indicates that the normally protective role of astrocytes may become deleterious in advanced disease.
Similar findings pointing to Purkinje neurons as playing a role have been reported for Guam disease or Amyotrophic lateral sclerosis / Parkinsonism-dementia complex [23]. As a whole, Purkinje cells appear to constitute one main target of neurodegeneration, not only in prion but also in prion-like diseases. Further studies focused on the interaction between this neuronal population and astroglia are being currently undertaken in order to elucidate this finding. Astrocytic ability to protect but also damage neurons might explain that the neurodegenerative progress is found closely associated to these glial cells.
Another notable observation here consists of the relationship between intensity and morphology observed in astroglia from the molecular layer. The intense radial gliosis observed in samples showing higher GFAP intensity suggests an advanced stage of the disease in agreement with conclusions provided in the Scrapie model [19]. Those samples showing a lower GFAP intensity showed a decrease of radial morphology while a different horizontal morphology appeared instead; they could correspond to an earlier stage of the disease based on the observations described in the study developed in the Scrapie model cited above. Cerebellar radial glia are used as ‘scaffolds’ guiding neuroprogenitor cells during development. This could suggest a glial stem cell response with the aim of compensating for neuronal loss, as previously proposed [9] on the basis of the evidence of that radial glial cell are important progenitor cells that contribute to gliogenesis. Furthermore, the relevance of studies developed in prion naturally affected animals is confirmed here as a reliable tool for helping to understand observations in affected human samples. Unlike in human disease, studies of Scrapie in sheep allow variable to be controlled and disease progress defined; the results of these studies may be extrapolated and provide useful information in regard to human disease. This association would predict what is happening in the neurodegenerative progress affecting humans.
By contrast, the involvement of microgliosis in AD affected samples differed greatly from that observed in CJD. Microglial activation appears to be less relevant in cerebellar tissue in AD than in CJD since reactive microglia were much more abundant in prion disease. However, other studies have indicated a higher significance for the role of microglia in AD. This could be explained due to the brain area studied or to the close relationship of this glial population with misfolded protein deposits or plaque - associated phagocytic activity, in prion disease [16] as well as in AD [24]. Thus, microglial activation is likely of greater significance in areas of brain wherein protein aggregates are found in AD, such as the hippocampus and cortex. Additional studies focused on further brain areas are planned in order to confirm this hypothesis.
The aggregation and spreading of aberrant proteins in prion and prion-like disease triggers a glial response. This glial response may itself impact the onset, progression, and severity of disease. To fully reveal the pathogenesis of prion and prion-like disease, there should be continued focus on both the astrocytic and microglial response to disease.
ACKNOWLEDGMENT
This work was funded by a grant from the University of Zaragoza (UZ2014-BIO-4). We thank to the Spanish brain banks from the Hospital Universitario Fundación Alcorcón and the Hospital Universitario Gregorio Marañón and Psiquiátrico de Ciempozuelos from Madrid, for providing brain samples.
REFERENCES
- Fernández-Borges N, Eraña H, Elezgarai SR, Harrathi C, Gayosso M, et al. (2013) Infectivity versus Seeding in Neurodegenerative Diseases Sharing a Prion-Like Mechanism. Int J Cell Biol 2013: 583498.
- Prusiner SB (2012) Cell biology. A unifying role for prions in neurodegenerative diseases. Science 336: 1511-1513.
- Prusiner SB (2013) Biology and genetics of prions causing neurodegeneration. Annu Rev Genet 47: 601-623.
- Abbott NJ, Rönnbäck L, Hansson E (2006) Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7: 41-53.
- Iadecola C, Nedergaard M (2007) Glial regulation of the cerebral microvasculature. Nat Neurosci 10: 1369-1376.
- Sofroniew MV (2009) Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci 32: 638-647.
- Perea G, Navarrete M, Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32: 421-431.
- Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119: 7-35.
- Barry D, Pakan JM, McDermott KW (2014) Radial glial cells: key organisers in CNS development. Int J Biochem Cell Biol 46: 76-79.
- Streit WJ, Kincaid-Colton CA (1995) The brain’s immune system. Sci Am 273: 54-55, 58-61.
- Graeber MB, Streit WJ (2010) Microglia: biology and pathology. Acta Neuropathol 119: 89-105.
- Burda JE, Sofroniew MV (2014) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81: 229-248.
- Beach TG, Walker R, McGeer EG (1989) Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia 2: 420-436.
- Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D (1989) Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol 24: 173-182.
- Diedrich JF, Bendheim PE, Kim YS, Carp RI, Haase AT (1991) Scrapie-associated prion protein accumulates in astrocytes during scrapie infection. Proc Natl Acad Sci USA 88: 375-379.
- Sikorska B, Liberski PP, Sobów T, Budka H, Ironside JW (2009) Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: a comparison with amyloid plaques in kuru, sporadic Creutzfeldt-Jakob disease and Gerstmann-Sträussler-Scheinker disease. Neuropathol Appl Neurobiol 35: 46-59.
- Ekdahl CT, Kokaia Z, Lindvall O (2009) Brain inflammation and adult neurogenesis: the dual role of microglia. Neuroscience 158: 1021-1029.
- Sarasa R, Martínez A, Monleón E, Bolea R, Vargas A, et al. (2012) Involvement of astrocytes in transmissible spongiform encephalopathies: a confocal microscopy study. Cell Tissue Res 350: 127-134.
- Hernández RS, Sarasa R, Toledano A, Badiola JJ, Monzón M (2014) Morphological approach to assess the involvement of astrocytes in prion propagation. Cell Tissue Res 358: 57-63.
- Bonifati DM, Kishore U (2007) Role of complement in neurodegeneration and neuroinflammation. Mol Immunol 44: 999-1010.
- Fukutani Y, Cairns NJ, Rossor MN, Lantos PL (1996) Purkinje cell loss and astrocytosis in the cerebellum in familial and sporadic Alzheimer’s disease. Neurosci Lett 214: 33-36.
- Sarasa R Junquera C, Toledano A, Badiola JJ, Monzón M (2015) Ultrastructural changes in the progress of natural Scrapie regardless fixation protocol. Histochem Cell Biol 144: 77-85.
- Sebeo J, Hof PR, Perl DP (2004) Occurrence of alpha-synuclein pathology in the cerebellum of Guamanian patients with parkinsonism-dementia complex. Acta Neuropathol 107: 497-503.
- Morgan D (2009) The role of microglia in antibody-mediated clearance of amyloid-beta from the brain. CNS Neurol Disord Drug Targets 8: 7-15.
Citation: Garcés M, Toledano A, Badiola JJ, Monzón M (2016) Morphological Changes of Glia in Prion and a Prion-Like Disorder. J Alzheimers Neurodegener Dis 2: 005.
Copyright: © 2016 Garcés M, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.