Multiplexed SNP Typing of DNA after Demineralization and Organic Extraction from a Bone
*Corresponding Author(s):
Zaorska KDepartment Of Histology And Embryology, University Of Medical Sciences, Poznan, Poland
Tel:+48 618546407,
Email:katarzyna.zaorska@gmail.com
Abstract
Analysis of DNA from bones and teeth still suffers from low efficiency of extraction methods and poor reliability of results during forensic typing, because of co-extraction of microbiological contaminants and DNA degradation.
Here we tested 15 variants of DNA extraction from old human bone using silica columns, organic phenol-chloroform-isoamyl alcohol solution, and combination of those two, with additional decalcification of different duration time. Multiplex PCR and single nucleotide polymorphism genotyping were performed to estimate the efficiency of extraction process and the level of DNA degradation. In comparison with silica-based method, the organic extraction gave more complete profiles with shorter time of decalcification, also less allele drop-outs and higher fluorescence level, independently of the additional cleaning. The minimal duration of successful decalcification was 48h. Decalcification is an essential step prior to DNA extraction from a bone. Phenol-chloroform- isoamyl alcohol solution effectively extracts DNA which can be further used in forensic analyses.
Keywords
DNA extraction; Decalcification; Forensic science; multiplex; Old bone; Single nucleotide polymorphism typing
INTRODUCTION
Positive identification of human fossil remains is a crucial issue in a forensic work. For over the last two decades multiplex STR (short tandem repeats) profiling has become a routine procedure in cases of identifying victims of mass disasters, missing persons and paternity testing [1]. Another issues are cases of remains with no presumable relatives or museum specimens and historical personages [2]. In such cases SNP (single nucleotide polymorphism) profiling has become one of the most relevant techniques and is often used as a supplementary tool in externally visible characteristics (EVCs), ancestry or kinship analyses [3, 4, 5]. Based on multiplex reactions using a single nucleotide genotyping techniques, it is possible to estimate complex traits such as eye color (IrisPlex tool) [6], hair color (HIrisPlex tool) mtDNA-based bio-geographic ancestry [7, 8] or use genomic DNA-based 52-plex assay for identification purposes [9]. Rapid and sensitive SNP technology often gives positive results for extremely degraded samples when conventional STR typing fails. Still, it faces many problems, e.g. allele drop-outs and incomplete or no amplification due to the high degree of sample degradation. DNA extraction process can itself increase other problematic issues, such as further DNA degradation, presence of inhibitors or loss of DNA template. Therefore, an adequate sample preparation and extraction method are crucial steps for successful forensic typing. Despite many studies and techniques an efficient DNA extraction from solid tissues (bones and teeth) can still constitute a significant problem. Specific morphological structure of a bone as well as variety of environmental conditions of bone preservation require peculiar handling with such a specimen [10]. There is usually a low level of endogenous DNA that is co-extracted with a large amount of exogenous microbiological DNA contamination from the surrounding of a bone, the bone itself or that is introduced during biological material collection and/or storage [11, 12]. Moreover, DNA extracted from old samples is degraded and the chemical reagents used during extraction process can cause further damage. Another problem is the presence of contamination and inhibitors in samples which co-extract with DNA and can affect an efficient amplification in the downstream PCR, STR or SNP profiling [13, 14]. However, sometimes bones are the only available specimen that can be the source of DNA. According to some authors, it is even better to extract DNA from bones or teeth than from poorly preserved soft tissue [15, 16]. It is also important what is considered an "old" material, since environmental conditions seem affect DNA preservation much stronger than the passage of time. Therefore, so far there is no universal protocol and distinct approaches are used depending on the stage of decomposition or time of the specimen's storage. Despite the fact that several commercial kits are designated to bones and teeth, still some authors choose more hazardous and complicated approaches or adapt kits designated to other tissues according to own lab experience and technical equipment.
Here we report the results of comparison of total of 15 different protocols for DNA extraction from an old bone. We used silica-based method, organic method using phenol- chloroform-isoamyl alcohol solution and combination of those two methods, with decalcification step of different duration prior to DNA extraction process. 6-SNP multiplex genotyping assay known as IrisPlex was used to verify the efficiency of DNA extraction process and estimate the level of DNA degradation and the possibility of successful amplification.
MATERIALS AND METHODS
Bone preparation
All steps of the preparation and extraction procedures were performed in the laminar flow cabinet. We used the diaphysis of ulna form human male, that was stored for over 50 years in The Department of Anatomy, University of Medical Sciences in Poznan, Poland, in moderate temperature and humidity conditions, in a wooden-glass cabinet. The bone was cut into several pieces, 4-cm long each. All pieces were subjected to mechanical and chemical cleaning. We used metal file to remove the outer layer of the bone and then immersed each bone piece in 10% sodium hypochlorite solution (NaOCl) (Sigma) for 15 min. Then, the bones were left to dry completely and washed in 70% ethanol and water, twice. After that, the bones were submerged in liquid nitrogen for about 10 min and crushed into small fragments. All fragments were grinded to a fine powder using a metal blender. If not used immediately, the bone powder was frozen and kept in -20?C until further analyses.
DNA Extraction
We performed 15 different variants of DNA extraction protocols. (Table 1) contains characterization of each variant of DNA isolation from bone tested in this study. One- and two-step extractions were carried out. During one-step extraction the bone powder was designated for the lysis and isolation process right after the bone was grinded. During two-step extraction we firstly applied the decalcification step (for 48h, 72h, 96h and 120h) using 0,5M EDTA (ethylene diamine tetra-acetic acid) solution, pH=8,0. For DNA isolation we used the silica-based method (QIAamp DNA Investigator Kit, Qiagen) and organic method, e.g. the Phenol-Chloroform-Isoamyl Alcohol (PCIA, 25:24:1) (Ambion) method. Additionally, the organic protocol was modified by adding the step of cleaning, using silica columns (Qiagen).
|
Sample ID |
Time of decalcification |
Method of DNA extraction |
Details |
Comments |
DNA concentration [ng/µl] |
|
S1 |
0 h |
PCIA |
a)Lysis buffer containing EDTA, DTT, SDS, NaCl; b)Precipitation performed with 3M Sodium Acetate and 99,8% ethanol. |
After 16h of lysis the bone sediment is still visible. |
30.7 |
|
S2 |
0 h |
PCIA + Silica |
29.4 |
||
|
S3 |
48 h |
PCIA |
After 16h of lysis the bone sediment is still visible, although the amount of the bone powder is smaller than in undecalcified samples. |
29.3 |
|
|
S4 |
48 h |
PCIA + Silica |
28.7 |
||
|
S5 |
72 h |
PCIA |
28.8 |
||
|
S6 |
72 h |
PCIA + Silica |
31.7 |
||
|
S7 |
96 h |
PCIA |
After 16h of lysis the pone powder dissolves completely. |
13.1 |
|
|
S8 |
96 h |
PCIA + Silica |
26.9 |
||
|
S9 |
120 h |
PCIA |
13.5 |
||
|
S10 |
120 h |
PCIA + Silica |
22.5 |
||
|
S11 |
0 h |
Silica |
c)The manufacturer's ATL lysis buffer; d)Precipitation performed with 99,8% ethanol. |
After 16h of lysis the bone sediment is still visible. |
5 |
|
S12 |
48 h |
Silica |
After 16h of lysis the bone sediment is still visible. |
46 |
|
|
S13 |
72 h |
Silica |
30.4 |
||
|
S14 |
96 h |
Silica |
After 16h of lysis the pone powder dissolves completely. |
3.2 |
|
|
S15 |
120 h |
Silica |
15.5 |
Table 1: Characterization of organic/silica-based methods of DNA extraction from a bone performed in this study. We tested an organic methods (using phenol-chloroform-isoamyl alcohol in ratio 25:24:1, respectively), silica-based method, and a combination of those two, with or without an additional pre-treatment step with 0,5M EDTA as a decalcifying agent for a specified period of time. Different chemical reagents were used for lysis of a bone powder and for DNA precipitation, as indicated. DNA yields were estimated using Nano Drop measurements. DTT-dithiothreitol; EDTA-ethylene diamine tetra-acetic acid; PCIA-phenol-chloroform-isoamyl alcohol; SDS- sodium dodecyl sulphate.
Decalcification Process
In order to decalcify the bone 5ml of 0.5M EDTA (Sigma) was used for every 1g of bone powder and the samples were incubated in conical tubes sealed with Para film, on a rotator mixer with slow rotation (10 rpm), at room temperature (RT), overnight. EDTA solution was changed every 24 hours by centrifugation (2000 g for 5 min), discarding the supernatant and adding the fresh EDTA. To test different variants, the bone powder was collected, respectively, after 48h, 72h, 96h and 120h of demineralization, and frozen in -20?C until the next step.
Silica Based Method
400mg of the bone powder of samples S11-S15 was incubated using the ATL lysis buffer from QIAamp DNA Investigator Kit and the protocol was followed according to the manufacturer's instructions.
Organic Method
We used self-prepared lysis buffer, containing 1mM dithiothreitol (DTT), 10mM Tris, 50mM EDTA (pH=8,0), 100mM NaCl and 0,5% sodium dodecyl sulphate (SDS). 1ml of the lysis buffer was used for every 500mg of the bone powder with addition of 30μl of proteinase K (20mg/ml, EURx). The lysis buffer was freshly prepared and the lysis was performed at 56?C in a thermoshaker with shaking (850 rpm), for 16h. After 16h of incubation 0,5ml of PCIA solution was added to all samples (S1-S10) and mixed by vortexing for 10 sec. Then, the samples were centrifuged for 15 min at 12 000 g at RT, to separate the layers. The upper DNA-containing aqueous phase of samples: S1, S3, S5, S7 and S9 was transferred to the new tubes and an equal volume of chloroform was added to the aqueous phase, mixed briefly and centrifuged at 12 000 g for 10 min at RT. This step was in order to remove any residual phenol and was proceeded twice. After the second centrifugation the upper phase was transferred to the new tubes and the samples were treated with 2,5μl of RNAse A (EURx) and incubated for 5 min at RT. The DNA was precipitated using 10% of the volume of 3M Sodium Acetate, pH=5,2-7,0, and one volume of ice cold 99.8% ethanol. The samples were centrifuged at full speed for 15 min at RT to pellet the DNA and washed with 200μl of ice cold 75% ethanol. The DNA was suspended in 50μl of nuclease-free water and left at 4?C overnight to dissolve.
Modified Organic Method
Samples S2, S4, S6, S8 and S10 were proceeded to the additional cleaning using silica columns (Qiagen). For this purpose, the lysis and centrifugation steps were performed as in the classic organic protocol (see above). Next, the upper aqueous layer was transferred to the new tubes, treated with 2.5μl of RNAse A and incubated for 5 min at RT. Then, the samples were proceeded to the Qiagen silica columns protocol.
The DNA concentration was estimated by the Nano-drop measurements. The reagent blank controls were prepared for each variant of extraction protocols.
DNA Analysis
All samples were genotyped using IrisPlex assay, which includes 6 SNPs in 6 genes. We performed multiplex PCR of those 6 variants followed by a minisequencing multiplex SNP typing using SNaPshot system. PCR and SBE (single base extension) primers were adopted from [17] and the SBE primers longer than 30bp were additionally HPLC purified (Oligo.pl). The first step of our analysis was the amplification of 6 fragments in one multiplex reaction. It was performed using 3μl of DNA extract, 1U of FastTaq polymerase (Roche), 200μM dNTP and defined concentration of each primer (Table 2), in a final volume of 10μl. Cycling conditions were as follows: 95 ºC for 10min, 33cycles of 95ºC for 13sec, 60 ºC for 30sec, 72 ºC for 30sec, and final extension 72 ºC for 15min. PCR products were treated with Exo/SAP mix containing 20U/μl of Exonuclease I and 1 U/μl of Shrimp Alkaline Phosphatase (New England BioLabs) to remove unincorporated dNTPs and primers. The conditions were 37 ºC for 60 min followed by 80 ºC for 15 min.
|
SNP ID |
Chromosome position |
Gene |
PCR primers (5' -> 3') |
PCR product |
Primer conc |
SBEprimer (5' -> 3') |
Primer direction |
Total primer length |
SBE primer conc |
Alleles detected |
|
rs1800407 |
15:25903913 |
OCA2 |
TGAAAGGCTGCCTCTGTTCT |
127 bp |
0,4 μM |
tttttttGCATACCGGCTCTCCC |
Forward |
23 bp |
0,1 μM |
G/A |
|
CGATGAGACAGAGCATGATGA |
||||||||||
|
rs16891982 |
5:33987450 |
SLC45A2 |
TCCAAGTTGTGCTAGACCAGA |
128 bp |
0,4 μM |
tttttttttttAAACACGGAGTTGA |
Forward |
29 bp |
1 μM |
G/C |
|
CGAAAGAGGAGTCGAGGTTG |
TGCA |
|||||||||
|
rs12203592 |
6:341321 |
IRF4 |
ACAGGGCAGCTGATCTCTTC |
115 bp |
0,4 μM |
tttttttttttttttaTTTGGTGGGTAA |
Forward |
35 bp |
0,3 μM |
C/T |
|
GCTAAACCTGGCACCAAAAG |
AAGAAGG |
|||||||||
|
rs12913832 |
15:26039213 |
HERC2 |
TGGCTCTCTGTGTCTGATCC |
87 bp |
0,4 μM |
ttttttttttttttttttttttttGCGTGCAG |
Reverse |
41 bp |
1,2 μM |
C/T |
|
GGCCCCTGATGATGATAGC |
AACTTGACA |
|||||||||
|
rs1393350 |
11:88650694 |
TYR |
TTCCTCAGTCCCTTCTCTGC |
80 bp |
0,4 μM |
tttttttttttttttttttttttaTTTGTAAA |
Reverse |
47 bp |
1,1 μM |
C/T |
|
GGGAAGGTGAATGATAACACG |
AGACCACACAGATTT |
|||||||||
|
rs12896399 |
14:91843416 |
SLC24A4 |
CTGGCGATCCAATTCTTTGT |
104 bp |
0,4 μM |
tttttttttttttttttttttttttttttaTCTTT |
Forward |
53 bp |
1,125 μM |
G/T |
|
CTTAGCCCTGGGTCTTGATG |
AGGTCAGTATATTTTGGG |
Table 2: Characteristics of 6 SNP markers used in the IrisPlex assay, including PCR and SBE primer sequences and concentrations, PCR-polymerase chain reaction; SBE-single base extension
The second step was a single base extension reaction performed in a total volume of 5μl using 2μl of ABI SNaPshot ready reaction mix (Applied Biosystems), 2μl of PCR product and defined concentration of each SBE primer under the following conditions: 95 ºC for 2min and 25 cycles of 95 ºC for 10sec, 50 ºC for 5sec and 60 ºC for 30sec. The products were cleaned using 1U/μl of Shrimp Alkaline Phosphatase (New England BioLabs), under conditions: 37 ºC for 60min followed by 80 ºC for 15min. 2μl of cleaned product was added to 7.75μl mix of formamide (Applied Biosystems) and 0.25μl of size standard (GeneScan 120 LIZ Size Standard, Applied Biosystems) and analyzed by the capillary electrophoresis on the ABI 3130 Genetic Analyzer (Applied Biosystems) using 36-cm length capillary and POP-7 polymer. Run parameters were as follows: injection voltage of 2.5 kV for 10sec and electrophoresis for 20min at 60 ºC. The results were analyzed with GeneMapper v4.0 (Figure 1).
Each sample was run total of three times. The reference samples were the buccal swab DNA from brown-eyed and blue-eyed Caucasian individuals (Figure1a, Figure1b). The amplicons for each SNP variant from 6-plex tested in this study were run in separate reactions to adjust the PCR conditions, primers’ concentrations and to evaluate the mobility of the SBE products in our genetic analyzer (not shown).
RESULTS AND DISCUSSION
The aim of our work was to determine the best method of DNA extraction from an old bone for the highest recovery of un degraded and amplifiable DNA. Most authors describe the efficiency of forensic DNA purification in the reference to the successful STR profiling. Here, we describe several variants of DNA extraction protocol from the old human bone to be tested in a multiplex single nucleotide polymorphism genotyping, commonly applied in routine forensic analyses. Based on the SNP profiles from all samples we assessed a consensus profile of tested DNA which includes all possible alleles that showed up in the three replicates in at least one sample. We discussed the efficiency of amplification in the reference to the presence or absence of expected alleles, as well as the fluorescence intensity measured as RFU (relative fluorescence unit) level. According to the standard STR typing, the level of 50RFU was taken as a threshold [18].
First of all, a rough mechanical cleaning of the outer surface of the bone, followed by a chemical cleaning, were performed. It has been previously shown that removing the outer surface of the bone is a critical step as it greatly reduces the amount of bacterial and/or fungal contamination [19-21]. Furthermore, sodium hypochlorite is a strong oxidant and, if used before isolation, it removes 99% of bacterial contamination as well as many inhibitors of downstream PCR analyses, leaving the endogenous DNA intact. Kemp & Smith [22] showed that immersion of a bone in at least 6% sodium hypochlorite for 15 minutes prior to DNA extraction was both the most practical and cost-effective approach for removal of exogenous contamination from ancient bones. In this study, the DNA concentration measurements were obtained using the Nano Drop, therefore the concentration value might constitute for both endogenous DNA as well as bacterial contamination. We performed series of amplification of different DNA extract volumes to determine the minimal DNA input for PCR (not shown). Only the 3μl or higher volume of DNA extract gave visible bands on the electrophoresis and that amount was used in further analyses in this study.
Apart from the contamination, specific composition of bone matrix, where DNA is bounded by hydroxyapatite to form aggregates, constitutes a problem during an extraction process and might require previous dissociation of DNA from this structure. It is still argued whether this step is necessary prior to DNA extraction. Some authors recommend incubation of the bone with 0.5M EDTA solution [23]. EDTA is used not only for decalcification, but also inactivates DNAses by chelating bivalent ions such as Mg2+ or Ca2+ [24]. However, the same authors have indicated that EDTA may cause further degradation of DNA. Moreover, it suggests that EDTA can itself act as an inhibitor in the downstream PCR. In our study, decalcification turned out to be an essential step for both silica-based and organic methods. We obtained a partial SNP profile for undecalcified sample (S11) in silica-based method, with high background and very low level of RFU under the threshold (Figure1c) and two other samples that were decalcified for 48h (S12) and 72h (S13) failed to amplify (Figure1d, Figure1e). Interestingly, those two samples gave the highest yields for silica-based method and no amplification could be the result of bacterial contamination of the samples or/and the presence of other inhibitors. Only the samples that were decalcified for 96h (S14) and 120h (S15) gave almost full profiles with only minor drop- outs: drop-out of the T allele in rs12203592 for sample S14 and drop-outs of the T alleles in rs12203592, rs1393350 and rs12896399 for sample S15 (Figure 1f, Figure 1g). The alleles of rs12203592 and rs1393350 were missing in most sample profiles in this study. Allelic drop outs are often encountered in forensic studies and are due to an under- representation of one of a pair of heterozygous alleles and preferential amplification of the other allele. This could be the result of DNA degradation or very small amount of endogenous DNA in the sample [25]. (Table 3) shows schematic combination of alleles in 6-plex profile tested in this study for all the samples.
|
consensus profile: |
G |
G |
C |
T |
C |
T |
C |
T |
G |
T |
||
|
marker ID: |
rs1800407 |
rs16891982 |
rs12203592 |
rs12913832 |
rs1393350 |
rs12896399 |
||||||
|
expected alleles: |
G |
A |
G |
C |
C |
T |
C |
T |
C |
T |
G |
T |
|
samples: |
|
|
|
|
|
|
||||||
|
S1 |
do |
- |
+ |
- |
do |
do |
+ |
+ |
do |
do |
+ |
do |
|
S2 |
do |
- |
+ |
- |
+ |
do |
+ |
+ |
+ |
do |
+ |
+ |
|
S3 |
do |
- |
+ |
- |
+ |
do |
+ |
+ |
+ |
+ |
+ |
+ |
|
S4 |
+ |
- |
+ |
- |
+ |
do |
+ |
+ |
+ |
do |
+ |
+ |
|
S5 |
do |
- |
do |
- |
+ |
+ |
+ |
+ |
do |
+ |
+ |
+ |
|
S6 |
do |
- |
+ |
- |
+ |
do |
+ |
+ |
+ |
do |
+ |
+ |
|
S7 |
sample failed to amplify |
|||||||||||
|
S8 |
do |
- |
+ |
- |
+ |
do |
+ |
+ |
+ |
do |
+ |
+ |
|
S9 |
+ |
- |
+ |
- |
+ |
do |
+ |
+ |
do |
do |
+ |
+ |
|
S10 |
do |
- |
+ |
- |
+ |
do |
+ |
+ |
+ |
do |
+ |
+ |
|
S11 |
do |
- |
+ |
- |
+ |
do |
+ |
+ |
do |
do |
do |
do |
|
S12 |
sample failed to amplify |
|||||||||||
|
S13 |
sample failed to amplify |
|||||||||||
|
S14 |
+ |
- |
+ |
- |
+ |
do |
+ |
+ |
+ |
+ |
+ |
+ |
|
S15 |
+ |
- |
do |
- |
+ |
do |
+ |
+ |
+ |
do |
+ |
do |
Table 3: Schematic combination of alleles present and absent from 6-plex profile tested in this study indicated for each sample variant of DNA extraction.
" +" equates to an allele present in the profile; " -" equates to an allele absent from the profile;
“do” equates to an allele drop out;
Similarly to the silica-based samples, the undecalcified sample (S1) in the organic method gave very poor profile with several drop outs, high background and RFU level mostly under the threshold (Figure 1h). However, the profile from the same sample, only with additional cleaning using the silica column (S2), turned out to be more complete with significantly higher fluorescence signal and almost no background noise (Figure 1i). This profile was similar to other samples in PCIA method, regardless of the time of decalcification (Figure 1j-1l). Only one sample (S7) failed to amplify (Figure 1m). Interestingly, cleaning the samples with silica columns in most cases gave the profile of similar alleles, only with relatively higher RFU and lower or no background noise (Figure1n-1q). Therefore, the application of decalcification seemed more crucial for the DNA recovery in our study, than the time of incubation with EDTA. Thus, we strongly recommend decalcification of a bone powder prior to DNA extraction for at least 48h. Likewise, Capelli & Tschentscher [26] suggested that decalcification of a bone is a necessary step with minimal duration of 48h, or longer.
It also performed a total demineralization of a bone using 15 ml of EDTA for every 1g of bone powder but the study lacks information about how long demineralization took place. What is more, the volume of EDTA exceeded 3 times the volume used in our study. Still, the authors obtained increased yields in comparison with the standard protocol, i.e. using EDTA only in lysis buffer overnight prior to PCIA extraction. On the contrary, other experiment showed no improvement of DNA extraction using total demineralization and DNA yields were even smaller when additional incubation with EDTA was preceded. It is hard to compare the influence of EDTA as decalcifying agent on DNA between the silica-based and organic methods tested in this study, as we did not obtain extension products for several samples (S7, S12, S13). Nevertheless, the fluorescence signal was higher for the sample profiles in PCIA method when compared to the silica-based method and the RFU values for the PCIA variants without additional cleaning were still higher than those for the silica columns only. Also, the yields in our study differed more between the extraction methods than between decalcified and undecalcified samples within one method. The exception were two samples (S12 and S13) that gave the higher yields for silica-based protocol but failed to give SBE products, which could indicate bacterial or other exogenous contamination or the presence of inhibitors in those samples.
It is worth mentioning, that the lysis buffer that we used for the organic protocol contained EDTA and that the bone powder was exposed to it for 16h during overnight incubation, even in case of samples that were not decalcified with pure EDTA solution. The exact composition of the manufacturer’s lysis buffer used in the silica-based method is not known, thus other than EDTA decalcifying agents might have been used. Another crucial problem, apart from the maximum DNA recovery, is potential presence of inhibitors that co-extract with DNA. When more bone powder is used the amount of inhibitors also increases, therefore it is more favorable to use less bone powder with more accurate and trusted extraction method. One approach is to use additional cleaning during the extraction process. Here, we applied the variant with purification the aqueous phase using the silica columns. It improved yields for 40% (2/5) of cleaned samples and diminished the background noise in all five (100%) SNP profiles.
Although the undeniable advantage of using the silica columns is the simplicity of technical handling and the safety of used reagents, the most efficient and reliable method in our study turned out to be the PCIA method. Similarly, some authors considered the phenol -chloroform method with pre-treatment with EDTA to be the best out of 3 methods tested, while others found both the silica and PCIA methods to be the finest for old bone out of 5 different methods tested. Moreover, the latter study related to bones dated to be about 750 years b.p. and the former considered bone as control sample for soft tissue DNA from cadaver of different stage of decomposition and the oldest sample was about 90 days old. What is even more interesting, demonstrated that the structural organization of crystallites and the overall lamellar structure is similar between modern and fossil bones, unless they are exposed to some detrimental environment conditions [27].
On the contrary, found the phenol-chloroform method, accompanied with total demineralization, not to be suitable for degraded specimens and for STR profiling, while others applied this protocol with successful STR typing, though with no decalcification step. One possible reason why the PCIA method worked best in our experiment is that we used the mixture of phenol-chloroform with isoamyl alcohol. Although phenol is commonly used for effective deproteinization, mixing it with chloroform and isoamyl alcohol in 25:24:1 ratio (respectively) prevents from preferential loss of some types of DNA [28]. The other reason in favor of PCIA in this study is the fact that, apart from ethanol, we used 3M sodium acetate (NaOAc) for DNA precipitation. Sodium acetate is used for removal of non-nucleic acid particles and making the DNA less soluble in a water phase, before precipitation with ethanol [29, 30]. Indeed, here, the NaOAc solution was used only in PCIA and not in silica-based protocol. Another interesting issue concerns using only the supernatant and/or the bone powder for extraction. It extracted supernatant only [31], whereas suggested that there is still some DNA left in the bone powder after the lysis. What is important, in our experiment, the samples that were exposed to EDTA incubation for 48h and 72h, were still in solid form after the lysis process, while samples decalcified for 96h and 120h dissolved completely during the lysis, regardless of the buffer (commercial or self- made). Thus, longer demineralization greatly facilitates the technical aspects of extraction especially using silica method, as even the small fragments of the bone powder can clog the column and prevent effective purification.
CONCLUSION
In conclusion, our experiment proves the PCIA method to be the most effective and reliable for DNA extraction from an old bone specimen, with the minimal of 48h of demineralization. Therefore, it can be preceded in a relatively short time of three working days and the DNA can be further used in downstream forensic analyses.
ACKNOWLEDGEMENT
The Ethics Committee of Poznan University of Medical Sciences approved the study protocol. The study was supported by the Poznan University of Medical Sciences grant for Young Scientists No. 502-14-02229373-09513.
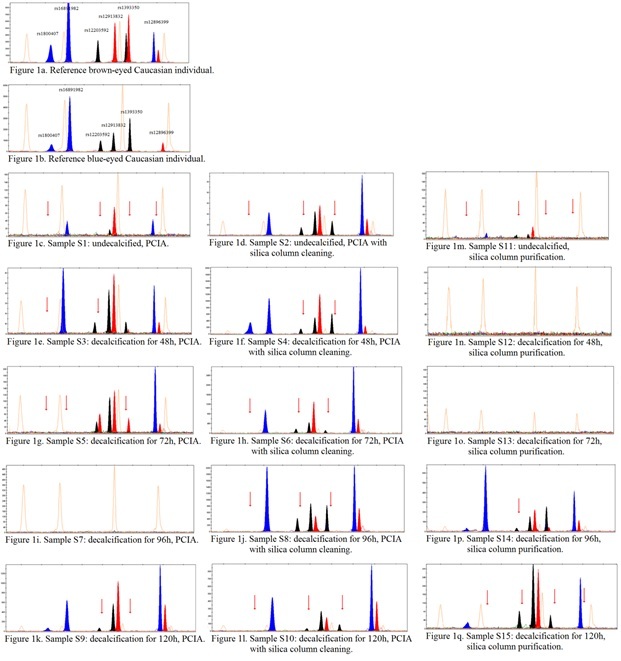 Figure 1: An assessment of IrisPlex assay’s profiles from 15 samples differing in the method of DNA extraction from a bone, from one individual. Red arrow indicates allele drop outs in the reference to the consensus sequence in this study; PCIA-phenol-chloroform-isoamyl alcohol;
Figure 1: An assessment of IrisPlex assay’s profiles from 15 samples differing in the method of DNA extraction from a bone, from one individual. Red arrow indicates allele drop outs in the reference to the consensus sequence in this study; PCIA-phenol-chloroform-isoamyl alcohol;
REFERENCES
- Oorschot R, Ballantyne KN, Mitchell RJ (2010) Forensic trace DNA: a review. Investig Genetics 1: 1-14.
- Kupiec T, Branicki W (2011) Genetic examination of the putative skull of Jan Kochanowski reveals its female sex. Croat Med J 52: 403-409.
- Wei YL, Qin CJ, Liu HB, Jia J, Hu L, et al. (2014) Validation of 58 autosomal individual identification SNPs in three Chinese populations. Croat Med J 55:10-13.
- Walsh S, Liu F, Wollstein A, Kovatsi L, Ralf A, et al. (2013) The HIrisPlex system for simultaneous prediction of hair and eye colour from DNA. Forensic Sci Int Genet 7: 98-115.
- Kayser M, De Kniff P (2011) Improving human forensics through advances in genetics, genomics and molecular biology. Nat Rev Genet 12:179-192.
- Kastelic V, Pospiech E, Draus-Barini J, Branicki W (2013) Prediction of eye color in the Slovenian population using the IrisPlex SNPs. Croat Med J 54: 381-386.
- Van Oven M, Vermuelen M, Kayser M (2011) Multiplex genotyping system for ancient inference of matrilineal genetic ancestry with continental resolution. Invest Genet 2:1-6.
- Endicott P, Metspalu M, Stringer C, Macaulay V, Cooper A, et al. (2006) Multiplexed SNP typing of ancient DNA clarifies the origin of Andaman mtDNA haplogroups amongst South Asian tribal populations. PLoS ONE 1:e81.
- Dario P, Oliveira AR, Ribeiro T, Porto MJ, Santos JC, et (2015) SNP for ID 52-plex in casework samples: “Cracking” bones and other difficult samples. Forensic Sci Int Genet 5: e118-e120.
- Parsons TJ, Weedn VW (1996) Preservation and recovery of DNA in postmortem specimens and trace samples. In: Forensic Taphonomy. The postmortem fate of human remains. CPR Press, New York, 109-138.
- Rohland N, Hofreiter M (2007) Ancient DNA extraction from bones and teeth. Nature Protocols 2:1756-1762.
- Von N, Harbeck M, Wiesbrock U, Schroeder I, Ritz-Timme S, et al. (2003) Extraction and amplification of nuclear and mitochondrial DNA from ancient and artificially aged bones. Leg Med (Tokyo) 5(Suppl): S169-S172.
- Loreille OM, Diegoli TM, Irwin JA, Coble MD, Parsons TJ, et al. (2007) High efficiency DNA extraction from bone by total demineralization. Forensic Sci Int Genet 1:191-195.
- Bouakaze C, Keyser C, Amory S, Crubezy E, Ludes B, et al. (2007) First successful assay of Y-SNP typing by SNaPshot minisequencing on ancient DNA. Int J Legal Med 121: 493-499.
- Piercy R, Sullivan KM, Benson N, Gill P (1993) The application of mitochondrial DNA typing to the study of white Caucasian genetic identification. Int J Leg Med 106: 85-90.
- Hagelberg E, Clegg JB (1991) Isolation and characterization of DNA from archeological
- Proc Biol Sci 244: 45-50.
- Walsh S, Lindenbergh A, Zuniga SB, Sijen T, De Knijff P, et al. (2011) Developmental validation of the IrisPlex system: Determination of blue and brown iris colour for forensic intelligence. Forensic Sci Int 5: 464-471.
- Hoff Olsen P, Mevag B, Staalstrom E, Hovde B, Egeland T, et al. (1999) Extraction of DNA from decomposed human tissue: An evaluation of five extraction methods for short tandem repeat typing. Forensic Sci Int 105: 171-183.
- Jakubowska J, Maciejewska A, Paw?owski R (2012) Comparison of three methods of DNA extraction from human bones with different degrees of degradation. Int J Legal Med 126: 173-178.
- Salamon M, Tuross N, Arensburg B, Weiner S, et al. (2005) Relatively well preserved DNA is present in the crystal aggregates of fossil bones. Proc Natl Acad Sci USA 102: 13783- 13788.
- Bogdanowicz W, Allen M, Branicki W, Lembring M, Gajewska M, et al. (2009) Genetic identification of putative remains of the famous astronomer Nicolaus Copernicus. Proc Natl Acad Sci USA 106: 12279-12282.
- Kemp BM, Smith DG (2005) Use of bleach to eliminate contaminating DNA from the surface of bones and teeth. Forensic Sci Int 154:53-61.
- Bender K, Schneider PM, Rittner C (2000) Application of mtDNA sequence analysis in forensic casework for the identification of human remains. Forensic Sci Int 133:103-107.
- Zoledziewska M, Gronkiewicz S, Dobosz T (2002) Comparison of various decalcificators in preparation of DNA from human rib bones. Anthropol Rev 65: 75-80.
- Hahn S, Garvin AM, Di Naro E, Holzgreve W (1998) Allele drop-out can occur in alleles differing by a single nucleotide and is not alleviated by preamplification or minor template increments. Genet Test 2: 351-355.
- Capelli C, Tschentscher F (2005) Protocols for ancient DNA typing. Methods in Molecular Biology. Clifton NJ 297: 265-278.
- Hofreiter M, Serre D, Poinar HN, Kuch M, Paabo S, et al. (2001) Ancient Nat Rev Genet 2: 353-359.
- Graham DE (1978) The isolation of high molecular weight DNA from whole organisms or large tissue masses. Anal Biochem 85: 609-613.
- Hasibe C, Tirpan AA (2010) Species determination of ancient bone DNA from fossil skeletal remains of Turkey using molecular techniques. Scientific Res Essays 5: 2250-2256.
- Cattaneo C, Smillie DM, Gelsthorpe K, Piccinini A, Gelsthorpe AR, et al. (1995) A simple method for extracting DNA from old skeletal material. Forensic Sci Int 74: 167-174.
- Hoss M, Paabo S (1993) DNA extraction from Pleistocene bones by a silica-based purification method. Nucleic Acid Res 21: 3913-3914.
Citation: Zaorska K (2020) Multiplexed SNP Typing of DNA after Demineralization and Organic Extraction from a Bone. Forensic Leg Investig Sci 6: 044.
Copyright: © 2020 Zaorska K, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.