Williams Syndrome - Review of Clinical Features and It’s Medical Considerations in Dental Treatment
*Corresponding Author(s):
Manikandan EkambaramDepartment Of Paediatric Dentistry, The University Of Hong Kong, 2nd Floor, Prince Philip Dental Hospital, 34-Hospital Road, Sai Ying Pun, Hong Kong
Tel:+852 28590260,
Fax:+852 25593803
Email:drmani@hku.hk
Abstract
Williams syndrome is a congenital disorder that involves multiple system. The condition is characterized by a number of developmental and physical abnormalities. The cause of this disorder is a micro deletion from the long arm of chromosome 7 at 7q11.23. The deleted portion of the chromosome 7q11.23 includes the ELN gene that codes for the structural protein elastin and therefore in the affected individuals arteries may become narrowed. Hypocalcaemia is also found in the affected individuals. Typical facial, oral, and dental features are characteristic of this syndrome. Presence of congenital heart defects and the risk of developing cardiac arrest make dental treatment challenging for such patients. This article narrates the etiology, pathogenesis, and clinical features including oral features of patients with Williams syndrome. A note on role of dental evaluation in diagnosis of Williams syndrome and medical consideration in the dental treatment of patients with Williams syndrome has also been presented.
Keywords
INTRODUCTION
Williams Syndrome (WS) was described by JCP Williams and AJ Beuren. This condition is also known by other names such as “Williams-Beuren syndrome”, “elfin-facies syndrome” and “idiopathic hypercalcemia-supravalvular aortic stenosis syndrome”. This is a congenital disorder characterized by several developmental and physical abnormalities [1-3]. The prevalence of this condition is 1 in 7,500 individuals [4]. Individuals affected with Williams syndrome has typical facial, and dental features that are relevant to dentists. Additionally, due to multisystem disorders including congenital cardiac defects the affected individuals pose a challenge for dental treatment. This article narrates the clinical features including oral, dental features and the significance of dental evaluation in the diagnosis of patients with Williams syndrome. The article also presents medical considerations in the dental treatment of patients with Williams syndrome.
ETIOLOGY
Williams Syndrome (WS) is caused by a micro deletion from the long arm of chromosome 7 at 7q11.23 [3]. The disorder is sporadic. However, an autosomal dominant inheritance pattern has also been established in which the affected individuals have a 50% chance of transmitting the disorder to their offspring. The disorder has no gender or ethnic predilection [1].
PATHOGENESIS
The deleted portion of the chromosome 7q11.23 includes the ELN gene that codes for the structural protein elastin, an essential component of the elastic fibres present in the connective tissue of several organs. The deletion of elastin explains few features of WS, such as part of the facial features, hoarse voice, bladder and bowel diverticula, cardiovascular disease, and orthopaedic problems. The pathogenesis of other features, such as hypercalcemia, mental retardation, and distinctive personality traits, are still unexplained. One possibility is that the loss of 1 or more genes contiguous to the ELN gene contributes to the phenotype. Since the elastin protein is an essential component of elastic fibres in the arterial wall, arteries (in general) become narrowed. Idiopathic infantile hypercalcemia is a feature of WS that can lead to severe irritability, vomiting, constipation, and muscle cramps associated with this condition [5]. Symptomatic hypercalcemia commonly does not manifest beyond childhood, but lifelong abnormalities of calcium and vitamin D metabolism may continue to exist. Hypercalciuria is common and predisposes to nephrocalcinosis. The reasons of the abnormality in calcium metabolism are unknown.
CLINICAL FEATURES
WS is a multisystem disorder. The cardiovascular system of affected individuals presents with supravalvular aortic stenosis, pulmonary artery stenosis, atrial septal defect, ventricular septal defect, aplasia of the portal vein, arterial hypertension and heart murmur. The affected individuals have mental deficiency with IQ between 41 and 80. Patients with this syndrome have typical facial features described as “Elfin face features” with reduced circumference of skull, craniosynostosis, broad forehead, short palpebral fissures, epicanthal folds, medial eyebrow flare, convergent strabismus, depressed nasal bridge, stellate pattern in iris, long philtrum, anteverted nostrils, wide mouth (Figure 1), drooping lower lip, hypoplastic mandible [6-8].
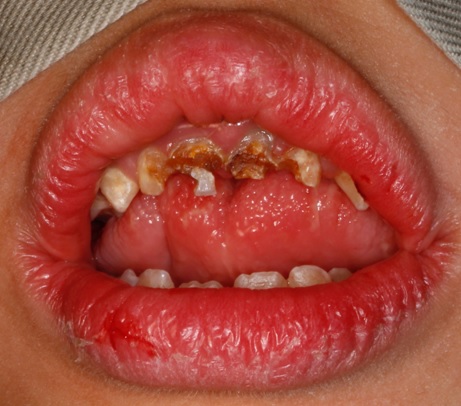 Figure 1: Extra-oral clinical picture of a patient with Williams syndrome presenting with prominent lips and wide mouth.
Figure 1: Extra-oral clinical picture of a patient with Williams syndrome presenting with prominent lips and wide mouth.
Hypercalcemiais not uncommon in the affected individuals and when present appears until the second year of life causing hypotonia and anorexia. The skeletal abnormalities include, growth deficiency (prenatal and postnatal), cranoisynostosis (secondary to microcephaly), retarded bone age, pectusexcavatum, hallux valgus and fifth finger clinodactyly [9]. Abnormalities of the renal system include renal anomalies, urinary tract infections, vesicoureter reflex, bladder diverticuli and nephrocalcinosis [10]. Constipation, peptic ulcer disease, cholelithiasis, diverticulitis, diabetes mellitus and obesity are the manifestations of gastrointestinal system. Other findings of this multisystem disorder include small penis, inguinal hernia, umbilical hernia.
GROWTH PATTERN
Williams Syndrome (WS) is associated with an abnormal growth pattern. The incidence of pre-natal growth deficiency is approximately 50-70% [5,11]. Feeding problems, prolonged colic, gastroesophageal reflex, and constipation lead to failure to thrive in 80% of infants with WS [12]. Until age four, weight gain and linear growth are poor. During childhood the rate of linear growth is 75% of normal [13]. In mid-childhood besides improvement in growth, 70% remain below the third percentile for mid parental height and are thus shorter than the children of similar age from their genetic background. Puberty often occurs early and is associated with a shorter growth spurt. These combined factors leads to an average final adult height that is below the third percentile [11].
SUPRAVALVULAR AORTIC STENOSIS IN WS
Supravalvular Aortic Stenosis (SVAS) is a rare but significantly characterized form of left ventricular outflow obstruction. The lesion involves the ascending aorta and may frequently be observed with associated pulmonary arterial stenosis or stenosis of other arteries, particularly at major branch points [14]. The onset and severity of disease varies but, if left untreated, may lead to heart failure, myocardial infarction, and death. Surgical treatment is the only option in symptomatic cases. Pathological studies of patients with SVAS recognized abnormalities in elastin structure and function. A quantitative deficiency of elastic fibres was also noted, resulting in an excess of smooth muscle clumps and bundles in the diseased media when compared with the normal aorta in which elastic fibres are in large number. The physiology of the vascular system, particularly the aorta, suggests that abnormalities in elastin could cause SVAS. Decreased vascular elasticity would have an adverse effect on vascular resistance and since hemodynamic stress is significant in the ascending aorta, this structure would be extremely affected [15].
RENAL ABNORMALITIES IN WS
The spectrum of abnormal renal findings ranged from minor abnormality such as bladder diverticula to major structural malformations such as renal agenesis or renal hypoplasia. The prevalence of renal agenesis is 0.2% in the normal population [16] and about 3.8% in WS patients. Burn [17] proposed the hypothesis that a delayed manifestation of renal diseases and urinary tract infections could be the result of hypercalcemia in early life. Like other arterial stenosis, incidence of stenosis in renal artery could be higher in WS. This hypothesis is supported by the findings that hypoplasia of the ascending aorta and the aortic loop and even long aortic stenosis was seen in a comparatively high number of patients [18]. Considering all the potential renal abnormalities sonographic examination of the kidneys and urinary tract should be included in the standard diagnostic work-up once the initial diagnosis of WS has been made.
HYPERCALCEMIA IN WS
The importance of hypercalcaemia related to WS has been a subject of debate; absolute hypercalcemia is routinely found only in infancy, if at all. Nearly all patients have symptoms of hypercalcemia in both infancy (vomiting, constipation, and irritability) and adult-hood (constipation, peptic ulcer disease, and frequent urination). Culler et al., [19] have suggested that an abnormality in calcitonin production or release could result in the relative hypercalcemia seen in WS. This relative hypercalcemia could result in an increased calcium load to the kidneys and in polyuria. The relationship of the abnormality in calcium metabolism to the pathogenesis of the other features of the syndrome is not known.
HYPERTENSION IN WS
Renal artery stenosis in WS has been correlated with hypertension [20]. Systolic blood pressure is raised in 25% of individuals with WS, diastolic blood pressure is raised in 9%, and both systolic and diastolic blood pressures are raised in 47% [21]. The presence of hypertension in WS increases with age. Cardiovascular disease accounts for nearly all of the morbidity and mortality of WS.
ORAL AND DENTAL FEATURES
Hypoplasia of the mandible, high-arched palate, macroglossia, frenulum hyperplasia, hypodontia, microdontia, abnormal incisor morphology, excessive inter dental spacing, anterior cross bite, anterior deep bite or open bite, small slender roots, dens invaginatus, hypoplastic bud-shaped maxillary primary second molars and mandibular permanent first molar, osteosclerotic changes in the lamina dura, delayed mineralization of the teeth, delayed eruption of teeth, hypoplastic defects of the primary and permanent dentition [6-9,22-24].
Hertzberg et al., [22] reported 40.7% abnormal incisor morphology in individuals with WS and microdontic maxillary lateral incisors (peg-shaped) were the most common. Thus, the altered incisor tooth morphology is an important dental feature of WS. Malocclusion occurs in majority of individuals with WS and this could possibly be due to the combined effects of hypodontia, tongue thrust, and connective tissue abnormality. Generalized widened periodontal ligament with vital teeth was recently reported [25] in a patient diagnosed with WS.
DIAGNOSIS
Diagnosis is generally made during mid-childhood when the typical facial features, cognitive profile, and cardiac findings become more apparent [1]. One of the first potential indicators that a neonate may have WS is “prolonged gestation”, classically more than 42 weeks. Other presentations are feeding difficulties, profuse vomiting, and failure to thrive. Due to variability in the clinical findings, diagnosis is routinely confirmed by chromosomal Fluorescent In-Situ Hybridization (FISH) analysis, detecting submicroscopic deletions of 7q11.23 [3] or by polymerase chain reaction [2]. Prenatal diagnosis of the deletion is obtainable for families at risk, using chorionic villous sampling or amniocentesis.
TREATMENT
Once a diagnosis of WS is made, the multisystem nature of the disorder requires continued treatment. Due to co-existence of multiple problems, many subspecialists including cardiologists, geneticists, ophthalmologists, neurologists, audiologists, dentist (with orthodontist), psychologists and occupational and physiotherapist may be involved in the care of these children. Psycho educational testing should be done routinely so that appropriate early intervention and school placement are provided. Whenever clinical symptoms appear in the urinary tract appropriate referral is indicated [5].
SUDDEN DEATH IN WS
Sudden death has been reported in many patients with WS. Significant, symptomatic obstruction may develop with any artery [5]. Coronary artery stenosis was found to have occurred in most of the cases that faced sudden death [26]. Abnormalities of the neurovasculature leading to stroke have also been reported [27].
SUGGESTIONS OF EARLY DIAGNOSIS
Early diagnosis depends on a careful assessment of infants who fail to thrive and are irritable, particularly when these factors are associated with a cardiac murmur, hernias, delayed developmental milestones, or facial features that are different from those of the parents or siblings [5]. In fact, the Committee on Genetics from the American Academy of Pediatrics has established a series of evaluations (Table 1) to assist the clinicians for the early diagnosis of WS [28].
| 1 | Complete physical and neurological examination |
| 2 | Growth parameters plotted on WS growth charts |
| 3 | Cardiology evaluation-Full clinical evaluation by a cardiologist with expertise and experience in pediatric patients that include 4-limb blood pressure measurements and echocardiography |
| 4 | Genitourinary system evaluation-Ultrasonography of bladder and kidneys, renal function studies (serum urea nitrogen and creatinine levels), urinalysis |
| 5 | Calcium determinations (serum calcium, spot urine calcium, and creatinine levels) |
| 6 | Thyroid function tests |
| 7 | Ophthalmologic evaluation |
| 8 | Multidisciplinary developmental evaluation (older than 2 years) |
| 9 | Fluorescence In Situ Hybridization (FISH) to determine ELN deletion |
Table 1: A series of evaluations recommended by the American Academy of Pediatrics for the early diagnosis of WS [28].
REASONS FOR DELAY IN DIAGNOSIS
Despite frequent visits to physicians for treatment of multiple medical problems, the diagnosis of WS is often delayed. The first reason for the delay is that the characteristic facial features may not be obvious at birth but become more obvious with time. Second, although the infants have symptoms that may be related to hypercalcemia, the symptoms are fairly nonspecific and serum calcium levels are not performed usually. Third, despite the fact that 80% of the children with WS have cardiac murmurs, most are asymptomatic in childhood and their murmurs are discharged as functional. Fourth, the developmental delays are often attributed to the chronic medical problems, and the small size and infantile facial features of children with WS make the patients look younger than their chronological age, thus further hide the developmental delays. Parents also may neglect to seek a diagnosis or may disagree with an early diagnosis for similar reasons. Morris et al., [5] describes that the affected children may seem to be “bright” because they are friendly, talkative, and “cute”.
THE ROLE OF DENTAL EVALUATION AND CEPHALOMETRIC ANALYSIS IN THE DIAGNOSIS OF WS
The clinical complexity WS is familiar, making it difficult to diagnose on the clinical picture, while genetic testing is not cheap and it is not cost effective to screen all patients. Therefore, dentists can be of help in diagnosing WS early. Tarjan et al., [29] performed Cephalometric analysis and dental examination of 33 patients with genetically confirmed diagnosis of WS. Cephalometric analysis of soft tissues presented that with normal SNA, SNB and ANB angles, the lips were in front of the line of harmony. This observation was present in all WS patients (n=33) but in none of the age-matched controls (n=100). No other variations were found between WS and control patients. This Cephalometric presentation is specific and sensitive for WS and can be applied in the diagnostic procedure.
MEDICAL CONSIDERATIONS IN THE DENTAL TREATMENT OF CHILDREN WITH WS
Cardiovascular and renal impairments tend to advance with age. Therefore, before performing any dental treatment, the extent of the disease should be assessed. Myocardial infarction or cardiac arrest has been reported after dental treatment under General Anesthesia (GA) in infancy [26]. Congenital heart defects usually appear in 53%-80% of the affected patients. This phenomenon requires both reduction of anxiety (from dental treatment) and administration of prophylactic antibiotics for bacterial endocarditis. In younger children, when bearing in mind the tendency to anorexia and vomiting, early dental examination and parental counseling are important. Preventive dental regimens and dietary counseling must be custom planned and implemented [30]. In children with WS, orthodontic assessment is generally recommended as soon as the patient’s co-operative level allows treatment [22]. Mucogingival problems resulting from anterior cross-bite and pseudo class III malocclusions should also be treated in primary and mixed dentition stages. The apprehension and hyperactive behaviour of children and adolescents with WS should be taken into consideration before planning any dental treatment. Seventy-five percent to 95% of children with WS have mild to moderate mental retardation, and about 80% of patients exhibit generalized anxiety disorder and are more likely to have Attention Deficit and Hyperactivity Disorder (ADHD). Large number (74%-95%) of children with WS is sensitive to sounds [31]. Oral, inhalational sedation should be considered for provision of less extensive dental treatment at a younger age to patients with WS as it can help to reduce anxiety and uncooperative behavior and thereby help the patient to cope with the procedure.
Treatment under GA is more appropriate for uncooperative older children who need extensive dental treatment. Prior consultation with a pediatrician is important when treatment under GA is considered, as there is a danger of an increase in blood pressure. Increased blood pressure can induce pulmonary edema, which is a life-threatening situation [32].
REFERENCES
- Lashkari A, Smith AK, Graham JM Jr (1999) Williams-Beuren syndrome: an update and review for the primary physician. Clin Pediatr (Phila) 38: 189-208.
- Kaplan P, Wang PP, Francke U (2001) Williams (Williams Beuren) syndrome: a distinct neurobehavioral disorder. J Child Neurol 16: 177-190.
- Francke U (1999) Williams-Beuren syndrome: genes and mechanisms. Hum Mol Genet 8: 1947-1954.
- Strømme P, Bjørnstad PG, Ramstad K (2002) Prevalence estimation of Williams syndrome. J Child Neurol 17: 269-271.
- Morris CA, Demsey SA, Leonard CO, Dilts C, Blackburn BL (1988) Natural history of Williams syndrome: physical characteristics. J Pediatr 113: 318-326.
- Friedman WF, Mills LF (1969) The relationship between vitamin D and the craniofacial and dental anomalies of the supravalvular aortic stenosis syndrome. Pediatrics 43: 12-18.
- Cohen MM (1976) Oral Facial Genetics Dysmorphic Syndromes with Craniofacial manifestations. Mosby, St Louis, USA.
- Goodman RM, Gorlin RJ (1977) Atlas of the face in genetic disorders. Williams syndrome. Mosby, St Louis, USA.
- Garabédian M, Jacqz E, Guillozo H, Grimberg R, Guillot M, et al. (1985) Elevated plasma 1,25-dihydroxyvitamin D concentrations in infants with hypercalcemia and an elfin facies. N Engl J Med 312: 948-952.
- Pankau R, Partsch CJ, Winter M, Gosch A, Wessel A (1996) Incidence and spectrum of renal abnormalities in Williams-Beuren syndrome. Am J Med Genet 63: 301-304.
- Pankau R, Partsch CJ, Gosch A, Oppermann HC, Wessel A (1992) Statural growth in Williams-Beuren syndrome. Eur J Pediatr 151: 751-755.
- Morris CA, Carey JC (1990) Three diagnostic signs in Williams syndrome. Am J Med Genet Suppl 6: 100-101.
- Jones KL, Smith DW (1975) The Williams elfin facies syndrome. A new perspective. J Pediatr 86: 718-723.
- Martin EC, Moseley IF (1973) Supravalvar aortic stenosis. Br Heart J 35: 758-765.
- Ewart AK, Jin W, Atkinson D, Morris CA, Keating MT (1994) Supravalvular aortic stenosis associated with a deletion disrupting the elastin gene. J Clin Invest 93: 1071-1077.
- Gruenwald P (1943) The normal changes in the position of the embryonic kidney. Anat Rec 85: 163-176.
- Burn J (1986) Williams syndrome. J Med Genet 23: 389-395.
- Wessel A, Pankau R, Kececioglu D, Ruschewski W, Bürsch JH (1994) Three decades of follow-up of aortic and pulmonary vascular lesions in the Williams-Beuren syndrome. Am J Med Genet 52: 297-301.
- Culler FL, Jones KL, Deftos LJ (1985) Imparied calcitonin secretion in patients with Williams syndrome. J Pediatr 107: 720-723.
- Deal JE, Snell MF, Barratt TM, Dillon MJ (1992) Renovascular disease in childhood. J Pediatr 121: 378-384.
- Kececioglu D, Kotthoff S, Vogt J (1993) Williams-Beuren syndrome: a 30-year follow-up of natural and postoperative course. Eur Heart J 14: 1458-1464.
- Hertzberg J, Nakisbendi L, Needleman HL, Pober B (1994) Williams syndrome--oral presentation of 45 cases. Pediatr Dent 16: 262-267.
- Torres CP, Valadares G, Martins MI, Borsatto MC, Díaz-Serrano KV, et al. (2015) Oral findings and dental treatment in a child with Williams-Beuren syndrome. Braz Dent J 26: 312-316.
- Cogulu D, Hazan F, Dindaroglu Fc (2015) Orofacial Findings and Dental Management of Williams Syndrome. Genet Couns 26: 437-442.
- Wong D, Ramachandra SS, Singh AK (2015) Dental management of patient with Williams Syndrome - A case report. Contemp Clin Dent 6: 418-420.
- Bird LM, Billman GF, Lacro RV, Spicer RL, Jariwala LK, et al. (1996) Sudden death in Williams syndrome: report of ten cases. J Pediatr 129: 926-931.
- Ardinger RH Jr, Goertz KK, Mattioli LF (1994) Cerebrovascular stenoses with cerebral infarction in a child with Williams syndrome. Am J Med Genet 51: 200-202.
- Committee on Genetics (2001) American Academy of Pediatrics: Health care supervision for children with Williams syndrome. Pediatrics 107: 1192-1204.
- Tarjan I, Balaton G, Balaton P, Vajo Z (2005) The role of dental evaluation and cephalometric analysis in the diagnosis of Williams-Beuren syndrome. Wien Klin Wochenschr 117: 226-228.
- Oncag A, Gunbay S, Parlar A (1995) Williams syndrome. J Clin Pediatr Dent 19: 301-304.
- Klein AJ, Armstrong BL, Greer MK, Brown FR 3rd (1990) Hyperacusis and otitis media in individuals with Williams syndrome. J Speech Hear Disord 55: 339-344.
- Moskovitz M, Brener D, Faibis S, Peretz B (2005) Medical considerations in dental treatment of children with Williams syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 99: 573-580.
Citation: Ekambaram M (2016) Williams Syndrome - Review of Clinical Features and It’s Medical Considerations in Dental Treatment. J Dent Oral Health Cosmesis 1: 003.
Copyright: © 2016 Manikandan Ekambaram, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.