Selective Toxicity and Angiogenic Inhibition by Euglenophycin: A Role in Cancer Therapy?
*Corresponding Author(s):
Paul V ZimbaDepartment Of Life Sciences, Center For Coastal Studies, Texas A&M University Corpus Christi, Texas, United States
Tel:+1 3618252768,
Fax:+1 3618252770
Email:Paul.Zimba@tamucc.edu
Abstract
A recently discovered euglenoid algal toxin, euglenophycin, is known to have anticancer properties. Euglenophycin is closely related to the alkaloid toxin solenopsin A, originally isolated from fire ants, that is an angiogenic inhibitor. Angiogenesis, the formation of new blood vessels, is required for the development of cancers. Three different human leukemia cell cultures (K562, THP-1, and Jurkat), a murine endothelial (SVR), and epithelial (IEC-6) cell lines were treated with euglenophycin at varying concentrations (0 - 100 μg/ml). After a 48 hour exposure, decreased production of Vascular Endothelial Growth Factor (VEGF) and Angiopoietin 2 (Ang-2) was observed using an Enzyme-Linked Immunosorbent Assay (ELISA). LC50: 48 hour for all cell lines was less than 50 μg/ml. Viability of cell cultures showed a dosage dependent decrease as compared to control using (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay and trypan blue exclusion. Morphological traits of SVR cells determined by phase contrast microscopy and scanning electron microscopy supported toxicity and viability findings. These data strongly suggest that euglenophycin possesses angiogenic inhibition properties, including metabolic retardation of VEGF and Ang-2 production and inhibition of cell proliferation against human leukemia and murine endothelial and epithelial cells. Future animal model research is required to assess euglenophycin’s potential as a new therapeutic anticancer treatment.
Keywords
INTRODUCTION
There are more than 100 different types of cancers that affect humans [1]. Cancers are generally classified via their tissue type of origin. Leukemia, originating from blood producing tissue or bone marrow, is one of the main types of cancer [2]. In people with leukemia, abnormal White Blood Cells (WBC) are produced from the bone marrow and released into the peripheral blood stream [2]. Unlike normal WBC, leukemias exhibit a higher rate of proliferation resulting in an accumulation of dysfunctional cells. Over time leukemia cells can crowd out normal red and white blood cells producing condition of anemia and hypoxia, which can stimulate the production of Vascular Endothelial Growth Factor (VEGF) [3], an important factor in angiogenesis.
Angiogenesis is the process of forming new blood vessels and is essential for tumor formation [4,5]. Paracrine signaling is the major method of cellular communication in the angiogenic process [6]. Endothelial cells that line the interior of all blood vessels are a source of numerous factors (stimulating/inhibiting) and corresponding target receptors that are involved in the angiogenic process including: VEGF-A, VEGF Receptor-2 (VEGFR-2), Angiopoientin-2 (Ang-2), and Tyrosine Kinase with Immunoglobulin-like and Endothelial Growth Factor-like Domains Receptor-2 (TIE2) [3,7,8]. It is widely accepted that cancerous cell masses have a critical size of ~1 mm3 and need new vasculature to grow larger [9,10]. Angiogenesis requires endothelial cell proliferation / migration and VEGF for success [11].
In mammals, the VEGF family consists of several members of signaling compounds (VEGF-A,-B,-C,-D,-E, and placenta growth factor). VEGF-A is a potent angiogenic signaling protein that regulates the differentiation, migration, proliferation, permeability, and survival of endothelial cells [12-14]. VEGF165 is the most abundantly expressed of the six isoforms of VEGF-A found in humans [11]. Of the three subtypes of cell surface receptor tyrosine kinases (VEGFR-1,-2, and -3), VEGFR-1 and VEGFR-2 bind to VEGF-A. Angiogenesis is thought to be mediated by VEGFR-2 binding and activation [11]. Within this study, VEGF165 will be examined and referred to as VEGF-A for brevity.
Historically cancer angiogenesis, which includes both types of angiogenesis, has been associated with solid tumors; however, newer evidence suggests that angiogenesis is also involved in leukemia formation and proliferation. Like growing tumor masses, leukemia cells depend on angiogenesis in the bone marrow for increased nutrient inflow and cellular waste removal [3].
Alkaloids from fresh water microorganisms, such as the toxin euglenophycin isolated from Euglena sanguinea Ehrenberg, may provide promising options for future novel cancer treatments [15]. This euglenoid species was the dominant algal species present in commercial freshwater ponds during a fish kill event [16], and chemical structure and various biological activities were later reported [15]. Euglenophycin has anticancer activity against HT-29 and HCT-116 cell lines derived from human colon cancer [15]. Interestingly, euglenophycin shares some degree of chemical structure similarity with the alkaloid solenopsin A, a potent angiogenic inhibitor [17]. Solenopsin A is a naturally occurring alkaloid isolated from the fire ant, Solenopsis invicta. Using the SVR cell proliferation bioassay, Arbiser et al., [17] found that solenopsin A can prevent the activation of angiogenesis through the Phosphatidylinositol-3-Kinase Pathway (PI3K).
HYPOTHESIS
This study examines cellular effects of euglenophycin exposure on human leukemias (THP-1, Jurkat, and K562), a modified murine endothelial cell line (SVR), and a murine non-cancerous epithelial cell line (IEC-6). We hypothesize that euglenophycin exposure to these cell lines will induce a significant change in the concentration of growth factor(s), the number of viable cells, cellular protusions, and cellular ultrastructure compared to control when treated for 48 hours. We further hypothesis that euglenophycin can serve as an effective treatment for certain cancers.
METHODS
Euglenophycin purification method
Euglenophycin was purified from the single celled euglenoid algae E. sanguinea Ehrenberg. At least seven strains of E. sanguinea have been identified as toxin producers (Zimba, pers. comm.). Cultures were grown in 20 L - 40 L batches in polycarbonate culture vessels using AF6 media [15]. Culture conditions were 20°C, 12:12 L:D cycle. The culture was filtered using a 7 μm mesh netting to collect a pellet of algal cells, then centrifuged and frozen at -80°C. The cell pellet was thawed and sonicated with methanol to extract toxin as previously described [15]. Euglenophycin was purified through mass-directed fractionation using an Agilent 1200 series HPLC-MS as previously described [15]. The purified euglenophycin was freeze dried (Labconco Freezone 4.5 L) and stored at -80°C. The toxin was reconstituted in Dimethyl Sulfoxide (DMSO) at a final treatment concentration not exceeding 0.1% total DMSO for cell assays.
Cell Cultures And Media Conditions
Human leukemia cell lines Jurkat (clone E6-1), K-562, and THP-1 were purchased from ATCC (Manassas, VA). Jurkat (clone E6-1) was established from a 14 year old male with acute T cell leukemia [18]. K-562 was established from a 53 year old female with chronic myelogenous leukemia [19]. THP-1 was established from a 1 year old male with acute monocytic leukemia [20]. All leukemia cell cultures were maintained in RPMI 1640 medium supplemented with 10% Fetal Bovine Serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml).
Murine endothelial cell line SVR (SVRN 1 ras) strain C57BL/6 was purchased from ATCC. The SVR cells were established from the pancreatic islets of Langerhans endothelium of adult mice. SVR cells were maintained in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) containing 4 mM L-glutamine, and supplemented with 10% Fetal Bovine Serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml).
Murine epithelial cell line IEC-6 was purchased from ATCC. IEC-6 cells were established from non-cancerous rat small intestine epithelium which synthesizes fibronectin and collagen, indicative of normally functioning epithelial cells [21]. IEC-6 were maintained in DMEM containing 4 mM L-glutamine, 1.5 g/L sodium bicarbonate, 4.5 g/L glucose and supplemented with 90% 0.1 U/ml bovine insulin, 10% Fetal Bovine Serum (FBS), penicillin (100 U/ml), and streptomycin (100 μg/ml).
All cell lines were incubated in T25 and T75 cell culture vented-capped flasks at 37°C and 5% CO2. Subculturing occurred two times a week as recommended. Cell integrity was monitored using a phase contrast inverted microscope for morphological assessment; trypan blue stain was used to assess cellular viability/mortality using a compound light microscope. Cell lines were grown for a minimum of one week before experimentation to ensure consistency of exponential cellular growth conditions. All experiments were performed on cell lines between passages 7 - 15 to prevent use of cells undergoing genetic drift or mutation.
Cell counts and viability
An improved Neubauer Bright-Line Hemacytometer was used with trypan blue stain to count viable and dead cells on a compound light microscope. Endothelial and epithelial adherent cell lines were washed with PBS then trypsinyzed using the Trypsin-EDTA Solution for Endothelial Cells 1X (Sigma, St. Louis, MO). When cells detached from the culturing flask, FBS in fresh RPMI 1640 media was used to inactivate the trypsin. Equal parts of homogenized cell-media mixture and trypan blue stain were combined for 2 - 5 minutes, and then cells were enumerated. For each treatment, counts were performed in triplicate and averaged.
Additionally, the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxy methyl phenyl)-2-(4-sulfophenyl)-2h-tetrazolium/ phenazinemethosulfate (MTS/PMS) assay called CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) was performed as a physiological method to detect viable respiring cells. After one hour of incubation a standard curve was created for each cell line, by plotting absorbance versus cell density. The normalization was necessary to account for differing metabolic activity between cell types and to establish lower and upper assay limits.
Euglenophycin treatment assays
At 50% - 70% confluency, cells were counted and seeded in a 24 well plate with 10,000 cells per well in 1 - 2 ml fresh media. Cells were grown overnight, exposed to 0, 3, 6, 9, 25, or 100 μg/ml of euglenophycin in triplicate for 48 hours, and viable cells were counted based on respiratory activity measured by the MTS assay. For these experimental conditions were used to minimize nutrient depletion during the 48 hour incubation period.
Light microscopy and scanning electron microscopy
Cells were counted using a Zeiss Axiolab compound light microscope. A Nikon TMS inverted microscope with phase contrast was used to observe all cell lines during maintenance and pre/post-experimentation. Phase contrast pictures of murine IEC-6 and SVR cells were taken with a mounted High Tech Computer (HTC) 5.0 megapixel digital camera on the inverted microscope at 400x magnification. Cells were in 24 well plates or T25 culture flask with appropriate growth medium when imaged. Images of controls and treatments were taken to compare morphological differences.
Cell surface morphology of the SVR endothelial cell line SEM was visualized using a modified protocol [22]. Glutaraldehyde fixative (50% EM grade) and 0.2 M Sorenson phosphate buffer pH 7.2 were obtained as a Karnovsky’s Fixative kit (Electron Microscopy Sciences). Glutaraldehyde and osmium tetroxide (dry; Electron Microscopy Sciences) were diluted with phosphate buffer to obtain 2% and 1% working solutions respectively.
Euglenophycin treatments were performed as described above with 12 mm diameter glass coverslips (Neuvitro Corporation) added to the well bottom. After 48 hours of exposure, media was aspirated, and coverslips were removed and placed in a glass petri dish containing 2% glutaraldehyde for 30 minutes to 1 hour at room temperature. Coverslips were carefully washed (three times) with phosphate buffer, then placed in a glass petri dish containing 1% osmium tetroxide for 30 minutes to 1 hour at room temperature. Coverslips were washed (three times) with phosphate buffer then placed in a dehydration gradient of 25%, 50%, 75%, 90%, and 100% ethanol for 5 - 10 minutes at each gradient step. After dehydration, coverslips were immediately dried using an Emitech (Ashford, England) K580 Critical Point Dryer, sputter coated with gold-palladium, and viewed using the JOEL NeoScope SEM.
SVR angiogenesis proliferation bioassay
The SVR angiogenesis proliferation bioassay was used to test for angiogenic inhibition of euglenophycin against endothelial cells as previously described [17]. The SVR cell line has been extensively used to test inhibition of angiogenesis [17,23-25]. SVR cells were treated with euglenophycin as described above. Carbazole was used as a positive control as it is a known angiogenic inhibitor [26]. After a 24 - 48 hour incubation period, media was aspirated and examined for non-attached dead cells. Wells were again trypsinized and cells counted after trypan blue staining using a hemocytometer. The SVR bioassay was repeated three times.
Enzyme-Linked Immunosorbent Assays (ELISA)
VEGF and Ang-2 concentrations were assayed using a human VEGF ELISA kit (Thermo Scientific, Waltham, MA) and a human Ang-2 ELISA kit (Invitrogen) in Jurkat, K-562, THP-1, and murine SVR cell lines. Using this assay, the minimum detectable concentrations of VEGF and Ang-2 are > 8.0 pg/ml and > 6.0 pg/ml, respectively. All treatments were performed in triplicate or greater according to manufacturer protocols. After a 48 hour incubation period absorbance values were obtained using a Thermo Scientific Multiskan Spectrum plate reader. A standard curve was generated from plotting the average absorbance for each standard versus the corresponding concentration. Blank wells containing only growth medium provided a baseline control.
Statistical approaches
The statistics software package JMP 9.0.2 (SAS Institute) was used for statistical analysis of experimental data. Data were examined for normalcy using the Shapiro-Wilk test (goodness-of-fit test) in addition to viewing normal quantile plots, histograms, and box blots. Data sets were transformed (log+1) if not distributed normally. One-way Analysis of Variance (ANOVA) was conducted to test significant differences among euglenophycin treatment means. Treatments having significant differences were compared using Tukey-Kramer HSD (α = 0.05). A 48 hour LC50 probit value was manually calculated. The Log10 values of euglenophycin treatment concentrations were calculated (n = 3). Percent mortality was calculated by the formula [(dead cells) / (live + dead cells)]*100. Abbott’s formula [(Treatment_dead-Control_dead) / (100– Control_dead)]*100 was used to obtain corrected percent mortality values. Corrected percent mortality was converted to probit values manually using Finney’s table [27]. Probit values versus Log10 of concentrations were graphed and a regression line was fitted. LC50:48hr was determined by solving for a Probit value (Y-value) of 5 (or 50%) then taking the inverse log of each concentration.
RESULTS AND DISCUSSION
Cell Viability
Human leukemia cell lines exhibited a dosage dependent response to euglenophycin concentration, with viable cells decreasing as euglenophycin concentration increased. Euglenophycin affected K562, THP-1, and Jurkat viable human leukemia cell numbers similarly, with 16 - 43 μg/ml euglenophycin treatment for 48 hours reducing cell numbers by 50% and 25 μg/ml euglenophycin causing over an 8-fold decrease in viability compared to control values. One hundred percent mortality occurred at 100 μg/ml. Significant differences in cell survival were evident from ANOVA analysis (Figure 1).
In this study, SVR modified endothelial cells were selected for euglenophycin treatment as a known assay for screening the anti-angiogenic potential of euglenophycin, a compound similar to solenopsin-A [17]. Euglenophycin inhibited the growth of ras-transformed SVR cells in a dosage dependent manner (Figure 2). Over 50% mortality was measured at 16 μg/ml euglenophycin with 100% SVR mortality at 100 μg/ml. Similarly, solenopsin-A reduced SVR cell number by 50% at 3 μg/ml as previously described [17]. Solenopsin-A is known to inhibit angiogenesis through the PI3K pathway [17] which suggests that euglenophycin may inhibit SVR cell proliferation. Arbiser et al. [17] found that only solenopsin-A was effective in inhibiting angiogenesis while other analogs of solenopsin with shorter carbon chains were not.
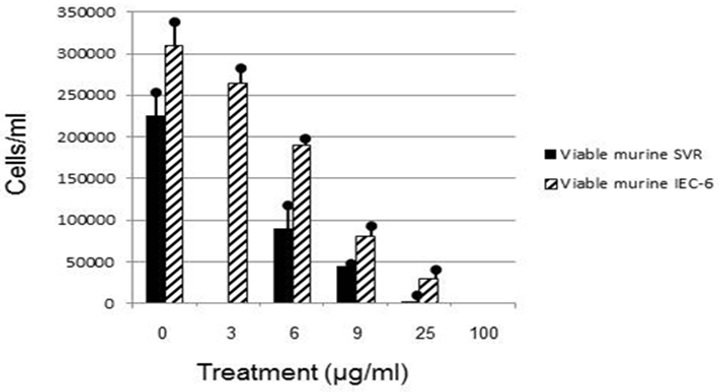
Figure 2: Viable murine SVR and murine IEC-6 cells after 48 hour euglenophycin exposure.
Euglenophycin LC50 in K562, THP-1, Jurkat, SVR, and IEC-6 cell cultures after 48 hour exposure ranged from 16-43 μg/ml. K562 leukemia had the highest cellular growth in the control treatment (Figure 3) which may explain the higher LC50 value of 43 μg/ml - higher division rates would compensate for cell mortality. The non-cancerous cell line IEC-6 had the 2nd highest LC50 of 40 μg/ml while the SVR endothelial cell line had the lowest at 16 μg/ml. These results suggest that SVR ras-transformed cells are more sensitive than IEC-6 cells to euglenophycin for growth inhibition, indicating euglenophycin’s potential as an anti-angiogenic therapeutic.
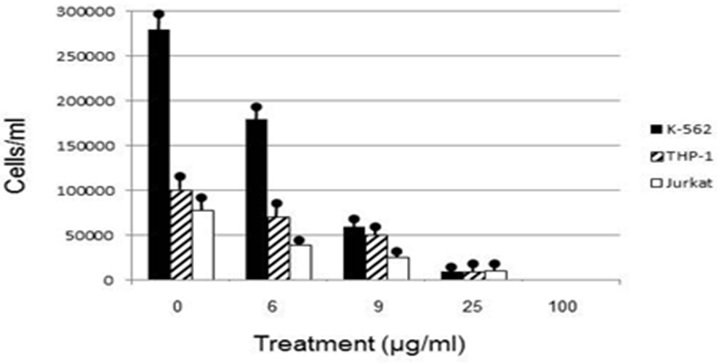
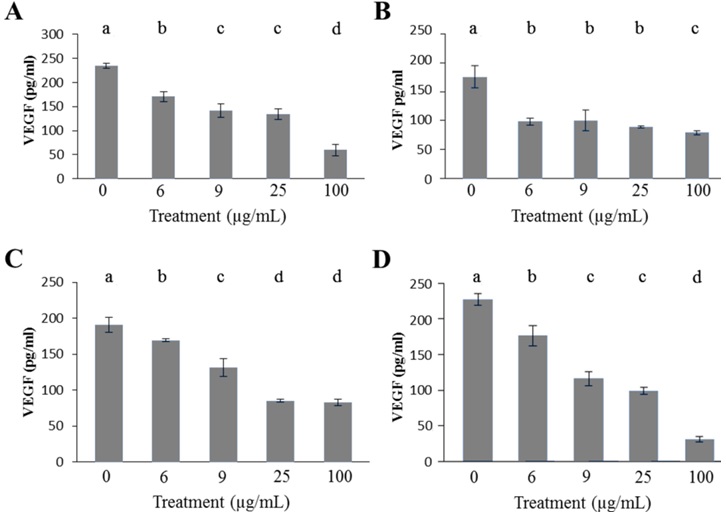
Figure 3: Ang-2 concentration determined by ELISA after a 48 hour euglenophycin exposurein K562 (A), THP-1 (B), and Jurkat (C) cell lines. In all treatments error bars represent the standard deviation of triplicate samples.
Morphological assessment via phase contrast and Scanning Electron Microscopy (SEM) provided further insight into euglenophycin’s toxicity on a cellular level. In this study, cellular protrusions are referred to as pseudopodia, as distinction between pseudopodia, filopodia, and lamellipodia were not made (exact location and distribution of cellular actin filaments and microtubules were not examined). Cell morphology was significantly affected by euglenophycin treatment. With increasing euglenophycin concentration, cell-to-cell contacts decreased and the pseudopodia stretched bi-directionally; at the highest concentrations, cellular rounding was observed (Figure 4). This suggests that euglenophycin alters endothelial surface communications, which require cell surface contact either through physical or ligand / receptor interactions. Euglenophycin’s exposure to SVR endothelial cells appears to immobilize them by inhibiting cellular protrusions. Endothelial migration is an essential component of angiogenesis which is orchestrated by cellular signals that are associated with cytoskeleton remodeling such as the PI3K pathway [28].
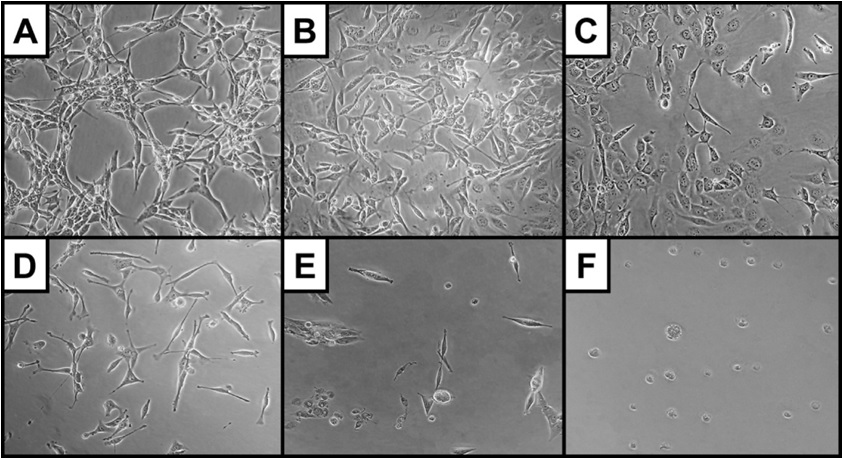
Figure 4: SVR cell morphology (at 400x magnification) after euglenophycin treatment (24 hrs) at 0 (A), 3 (B), 6 (C), 9 (D), 25 (E), and 100 µg/mL (F). Note the increased rounding of cells and decreased cell: cell contact at higher concentrations.
Via SEM it was apparent that microvilli were present on the squamous non-treated cells but decreased with higher concentrations of euglenophycin treatment. At dosages >25 μg/ml, the sphere-like cell surfaces appeared blebbed or furrowed and not entirely smooth. SVR microvilli and pseudopodia may be acting as antennae on the cellular protrusions for the endothelial cells much like an endothelial tip cell [29,30]. Similar observations were made in a study of spontaneous hypertensive rat endothelial cells in which numerous microvilli were present on the apical surface [31]. Lamalice et al., [28] also states that endothelial cell migration is partially driven by chemotaxis, in which VEGF and angiopoietin gradients are detected by membrane protrusions (filopodia).
Effect Of Euglenophycin On VEGF And ANG-2 Concentration
Leukemia, as with other cancer, proliferates within complex microenvironments in which cells have increased levels of growth factors such as VEGF and Ang-2 [3,7,14].These growth factors are important steps in the angiogenic process of forming new blood vessels. Most cells involved in the angiogenic process are non-cancerous; however, they assist in forming new vasculature supporting uncontrolled cancer growth. Figure 2 shows VEGF concentration for human K562, THP-1, Jurkat, and murine SVR cell cultures after a 48 hour euglenophycin exposure period. With a 6 μg/ml euglenophycin treatment, all leukemias had an 11% - 44% decrease in VEGF concentration. SVR cells experienced a 22% decrease as compared to control. At concentrations of 9-100 μg/ml, all cells showed a reduction in VEGF concentration after toxin exposure; however, the response was not dosage dependent and some treatment values were not significantly different from each other.
All leukemia cells exhibited a similar Ang-2 response to euglenophycin. Non-treated leukemia cells had Ang-2 concentrations from 653 pg/ml to 1721 pg/ml (Figure 3). K562 cells experienced a 58% decrease in Ang-2 concentration as compared to control after a 6 μg/ml euglenophycin treatment. At 100 μg/ml euglenophycin, all leukemia cell lines had Ang-2 levels of 78 pg/ml - 238 pg/ml. Significant differences in Ang-2 concentration were evident from ANOVA analysis for the three cell lines (Figure 3).
Interestingly, all leukemias showed detectable concentrations of VEGF and Ang-2 for 100 μg/ml euglenophycin treatments while each well had no detectable viable cells present. One explanation is the half-life of VEGF is 6-8 hours under hypoxic conditions [32]. The leukemia cells could have produced the VEGFs then died. Another possible explanation is that not all the background absorbance was removed prior to measuring ELISA absorbance results.
Qualitative analysis of VEGF and Ang-2 provides insight into euglenophycin’s effect on SVR cells (Figure 3). A reduction in VEGF concentration occurred with increased euglenophycin concentration, which also caused SVR endothelial cells to have fewer cellular protrusions and microvilli.
Murine SVR ultrastructure was examined by SEM after euglenophycin exposure (0, 3, 6, 9, 25, and 100 μg/ml dosage) for 48 hours (Figure 5). Pseudopodia loss occurs with increasing concentrations of euglenophycin. Microvilli were present on the squamous non-treated cells but decreased with higher concentrations of euglenophycin. SVR cells dosed with euglenophycin (0, 3 and 6 μg/ml concentration) had loss of ultrastructure as euglenophycin concentrations increase, which was more apparent when comparing control to 25 μg/ml euglenophycin treated endothelial cells. At 25 μg/ml euglenophycin, the SVR cell surface appeared smooth and without microvilli. Similar to phase contrast observations, a noticeable rounding of cells and pseudopodia loss was observed at higher euglenophycin concentrations. At 100 μg/ml, spherical shaped cells had no defined ultrastructure, such as pseudopodia and microvilli, as compared to control. The sphere-like cell surfaces appear blebbed or furrowed and not entirely smooth.
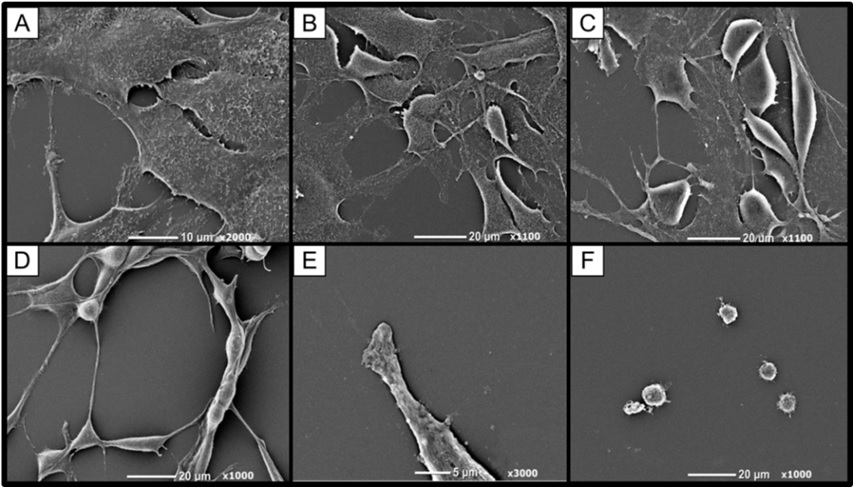
Figure 5: Murine SVR endothelial cell morphological changes after 48 hour euglenophycin exposure as determined by SEM. SVR cells exposed to (A) 0, (B) 3, (C) 6, (D) 9, (E) 25, and (F) 100 μg/ml toxin, respectively. As concentration of euglenophycin increases, microvilli loss increases and cell bodies begin to round in shape with less pseudopodia present.
A cell’s surface area is reduced when the number of cellular protrusions and microvilli decrease, which presumably causes a concomitant reduction in the number of cell surface receptors, such as VEGFR-2 and Ang-2. Reducing the paracrine signaling of VEGF between endothelial cells would assist in the inhibition of the angiogenic process through immobilization of endothelial cells.
In summary, euglenophycin can reduce the number of viable leukemia cells and reduce leukemic metabolic activity in vitro. Having a reduced number of viable leukemia cells, which produce less pro-angiogenic growth factors, is desirable in cancer therapies. Euglenophycin’s ability to inhibit SVR growth and its similar chemical structure to solenopsin-A suggest that future studies could help determine if euglenophycin acts to inhibit angiogenesis through the PI3K pathway. Evidence herein of euglenophycin’s ability to inhibit angiogenesis in vitro should stimulate future research in an animal model.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
References
- National Institutes of Health, What Is Cancer? National Cancer Institute, Maryland, USA.
- National Institutes of Health, What You Need To Know About Leukemia. National Cancer Institute, Maryland, USA.
- Trujillo A, McGee C, Cogle CR (2012) Angiogenesis in acute myeloid leukemia and opportunities for novel therapies. J Oncol 2012: 128608.
- Folkman J (1971) Tumor angiogenesis: therapeutic implications. N Engl J Med 285: 1182-1186.
- Folkman J (2007) Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov 6: 273-286.
- Tsou R, Fathke C, Wilson L, Wallace K, Gibran N, et al. (2002) Retroviral delivery of dominant-negative vascular endothelial growth factor receptor type 2 to murine wounds inhibits wound angiogenesis. Wound Repair Regen 10: 222-229.
- Maffei R, Martinelli S, Castelli I, Santachiara R, Zucchini P, et al. (2010) Increased angiogenesis induced by chronic lymphocytic leukemia B cells is mediated by leukemia-derived Ang2 and VEGF. Leuk Res 34: 312-321.
- Felcht M, Luck R, Schering A, Seidel P, Srivastava K, et al. (2012) Angiopoietin-2 differentially regulates angiogenesis through TIE2 and integrin signaling. J Clin Invest 122: 1991-2005.
- Weidner N, Semple JP, Welch WR, Folkman J (1991) Tumor angiogenesis and metastasis--correlation in invasive breast carcinoma. N Engl J Med 324: 1-8.
- Siemann DW, Chaplin DJ, Horsman MR (2004) Vascular-targeting therapies for treatment of malignant disease. Cancer 100: 2491-2499.
- Holmes K, Roberts OL, Thomas AM Cross MJ (2007) Vascular endothelial growth factor receptor-2: structure, function, intracellular signalling and therapeutic inhibition. Cell Signal 19: 2003-2012.
- Dias S, Shmelkov SV, Lam G, Rafii S (2002) VEGF(165) promotes survival of leukemic cells by Hsp90-mediated induction of Bcl-2 expression and apoptosis inhibition. Blood 99: 2532-2540.
- Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro Oncol 7: 452-464.
- Schuch G, Machluf M, Bartsch G Jr, Nomi M, Richard H, et al. (2002) In vivo administration of Vascular Endothelial Growth Factor (VEGF) and its antagonist, soluble neuropilin-1, predicts a role of VEGF in the progression of acute myeloid leukemia in vivo. Blood 100: 4622-4628.
- Zimba PV, Moeller PD, Beauchesne K, Lane HE, Triemer RE (2010) Identification of euglenophycin--a toxin found in certain euglenoids. Toxicon 55: 100-104.
- Zimba PV, Rowan M, Triemer R (2004) Identification of euglenoid algae that produce ichthyotoxin(s). J Fish Dis 27: 115-117.
- Arbiser JL, Kau T, Konar M, Narra K, Ramchandran R, et al. (2007) Solenopsin, the alkaloidal component of the fire ant (Solenopsis invicta), is a naturally occurring inhibitor of phosphatidylinositol-3-kinase signaling and angiogenesis. Blood 109: 560-565.
- Schneider U, Schwenk HU, Bornkamm G (1977) Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int J Cancer 19: 621-626.
- Lozzio CB, Lozzio BB (1975) Human chronic myelogenous leukemia cell-line with positive Philadelphia chromosome. Blood 45: 321-334.
- Tsuchiya S, Yamabe M, Yamaguchi Y, Kobayashi Y, Konno T, et al. (1980) Establishment and characterization of a human acute monocytic leukemia cell line (THP-1). Int J Cancer 26: 171-176.
- Quaroni A, Wands J, Trelstad RL, Isselbacher KJ (1979) Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol 80: 248-265.
- Passey S, Pellegrin S, Mellor H (2007) Scanning electron microscopy of cell surface morphology. Curr Protoc Cell Biol 4: 17.
- Arbiser JL, Moses MA, Fernandez CA, Ghiso N, Cao Y, et al. (1997) Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc Natl Acad Sci USA 94: 861-866.
- Arbiser JL, Klauber N, Rohan R, van Leeuwen R, Huang MT, et al. (1998) Curcumin is an in vivo inhibitor of angiogenesis. Mol Med 4: 376-383.
- Bai X, Cerimele F, Ushio-Fukai M, Waqas M, Campbell PM (2003) Honokiol, a small molecular weight natural product, inhibits angiogenesis in vitro and tumor growth in vivo. J Biol Chem 278: 35501-35507.
- Arbiser JL, Govindarajan B, Battle TE, Lynch R, Frank DA, et al. (2006) Carbazole is a naturally occurring inhibitor of angiogenesis and inflammation isolated from antipsoriatic coal tar. J Invest Dermatol 126: 1396-1402.
- Finney DJ (1952) Probit Analysis. (2ndedn), Cambridge University Press, Cambridge, UK.
- Lamalice L, Le Boeuf F, Huot J (2007) Endothelial cell migration during angiogenesis. Circ Res 100: 782-794.
- Adams RH, Alitalo K (2007) Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8: 464-478.
- Herbert SP, Cheung JY, Stainier DY (2012) Determination of endothelial stalk versus tip cell potential during angiogenesis by H2.0-like homeobox-1. Curr Biol 22: 1789-1794.
- Hazama F, Ozaki T, Amano S (1979) Scanning electron microscopic study of endothelial cells of cerebral arteries from spontaneously hypertensive rats. Stroke 10: 245-252.
- Shima DT, Deutsch U, D’Amore PA (1995) Hypoxic induction of Vascular Endothelial Growth Factor (VEGF) in human epithelial cells is mediated by increases in mRNA stability. FEBS Lett 370: 203-208.
Citation: Citation: Zimba PV, Ordner P, Gutierrez DB (2016) Selective toxicity and angiogenic inhibition by euglenophycin a role in cancer therapy? J Cancer Biol Treat 3: 008.
Copyright: © 2016 Paul V Zimba, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.