Journal of Obesity & Weight Loss Category: Medical
Type: Research Article
Current Advances in Pathogenesis in Obesity: Impact of Hypothalamic Glioses
*Corresponding Author(s):
Kulvinder Kochar KaurKulvinder Kaur Centre For Human Reproduction, Panjab University, Panjab, India
Tel:+ 91 9501358180,
Fax:91 181 4613422
Email:kulvinder.dr@gmail.com
Received Date: Jun 26, 2017
Accepted Date: Jan 10, 2018
Published Date: Jan 26, 2018
Abstract
Obesity has assumed epidemic proportions worldwide. Still though some therapies are approved by Food and Drug Administration (FDA) like Qsymia (topiramate and phentermine), contrive (naltrexone and bupropion), lorcaserin, all are not available in all countries, besides the use of GLP1 receptor agonist liraglutide for obesity as well besides diabetic control. Whatever one has to offer causes very little weight loss besides maintenance of weight loss despite lifestyle interventions is a herculean task as shown by Diabetes Prevention Program (DPP) and Look AHEAD studies. The only effective therapy remains bariatric surgery which cannot be offered to all with its cost, complications. Hence more in-depth knowledge is needed regarding pathogenesis of obesity. Here we review further how high fat diet effects gut flora through effects of TLR4 signaling with help of intermediates like Myeloid Differentiation factor (MyD88) and TRIF. Besides the role of hypothalamic inflammation occurring as a primary event in High Fat Diet (HFD) has been emphasized and how hypothalamic glioses affects with neuronal injury occurring as a primary event and ultimate damage of Proopiomelanocortin (POMC) neurons which changes ratio of orexigenic Agrp (Agouti Related Peptide) to POMC neurons ratio in favor of weight gain. If one can intervene at hypothalamic inflammation level newer therapies can be generated.
Keywords
Gliosis; Gut immunity; Gut micro biota; HFD; Hypothalamic inflammation
ABBREVIATIONS
FDA: Food and Drug Administration
POMC: Proopiomelanocortin
AgRP: Agouti Related Peptide
WAT: White Adipose Tissue
HFD: High Fat Diet
CCL2: C-C motif chemokine Ligand 2
TLR: Toll Like Receptor
IR: Insulin Resistance
MyD88: Myeloid Differentiation primary response 88
DIO: Diet-Induced Obesity
GF: Germ Free
JNK: c-Jun N-terminal Kinase
NFKB: Nuclear Factor Kappa B
SOCS3: Suppressor of Cytokine Signaling 3
PTB1B: Protein Tyrosine Phosphatase 1B
EPA: Eicosapentaenoic Acid
DHA: Docosahexaenoic Acid
ER: Endoplasmic Reticulum
GPR120: G-Protein coupled Receptor 120
ICV: Intracerebroventricular
HI: Hypothalamic Inflammation
POMC: Proopiomelanocortin
AgRP: Agouti Related Peptide
WAT: White Adipose Tissue
HFD: High Fat Diet
CCL2: C-C motif chemokine Ligand 2
TLR: Toll Like Receptor
IR: Insulin Resistance
MyD88: Myeloid Differentiation primary response 88
DIO: Diet-Induced Obesity
GF: Germ Free
JNK: c-Jun N-terminal Kinase
NFKB: Nuclear Factor Kappa B
SOCS3: Suppressor of Cytokine Signaling 3
PTB1B: Protein Tyrosine Phosphatase 1B
EPA: Eicosapentaenoic Acid
DHA: Docosahexaenoic Acid
ER: Endoplasmic Reticulum
GPR120: G-Protein coupled Receptor 120
ICV: Intracerebroventricular
HI: Hypothalamic Inflammation
INTRODUCTION
In our previous reviews we have emphasized on the etiopathogenesis of obesity and further on nutrient metabolism where we emphasized on the role of how high fat diet interacts with gut flora along with the role in hypothalamic inflammation [1,2]. Here we further explore on the role of HFD in White Adipose Tissue (WAT) inflammation and mechanism of glioses as being important for the development of hypothalamic inflammation and role in free fatty acid metabolism [1,2]. Both human and animal studies now help in explaining how hypothalamic inflammation precedes the development of peripheral inflammation known for over 2 decades now and although the role of gut microbiota has been emphasized here, Caesar et al., highlight how with changing fish oil diet in high fat diet fed mice gut flora gets altered to helpful Lactobacillus and Akkermansia muciniphilia strains and further role of gut anti-inflammatory agents is highlighted in therapeutic control of obesity.
HIGH FAT DIET (HFD) AND WHITE ADIPOSE TISSUE (WAT) INFLAMMATION
Role of HFD in Obesity
Diets rich in saturated dietary lipids are associated with increased WAT inflammation and metabolic diseases, where as diets rich in polyunsaturated fatty acids have been shown to counteract inflammation and promote a lean and metabolically healthy phenotype [3-6]. Host diet also has a major influence on gut microbial phenotype and changes in the gut microbiota and thereby host physiology [7,8]. Studies showing that Germ Free (GF) mice are protected against diet induced obesity exhibit decreased WAT inflammation and Insulin Resistance (IR) have lead to suggestions that microbial factors may directly contribute to WAT inflammation along with adverse metabolic sequences [9-12]. In healthy humans and mice circulating factors from microbiota have been found [13]. Furthermore an increased influx of microbial factors has been linked to inflammation and impaired glucose metabolism through the activation of Toll Like Receptor (TLR) dependent signaling [14,15]. Various genetic mouse models have shown that deletion of components of the TLR signaling pathway is associated with protection against WAT inflammation and/or rescue of metabolically perturbed phenotypes [16]. However although TLR ligands may be of bacterial origin they may also come from diet or the host, and thus gnotobiotic models are required to determine how the gut microbiota contributes to WAT inflammation and upon diet change thus Caesar et al., aimed to determine whether WAT inflammation induced by dietary lipids is mediated through the gut microbiota and to identify molecular mechanisms via which gut microbiota induce macrophage accumulation in WAT [17]. They showed that mice fed lard for 11 weeks have increased TLR activation and WAT inflammation and reduce insulin sensitivity as compared to mice fed fish oil and further phenotypic differences in microbiota composition. Trif-/- and MyD88-/- mice are protected against lard induced WAT inflammation and insulin sensitivity. Experiments in GF mice showed that an interaction between gut microbiota and saturated lipids promote WAT inflammation independent of adiposity. They also showed that the chemokine CC motif ligand 2 (CCL2) is the one which adds to microbiota induced WAT inflammation in lard fed TLR signaling upon challenging with a diet rich in saturated lipids [18].
Role of gut microbiota
They showed that the type of dietary lipids affects the gut microbiota and that the gut microbiota further contributes to the phenotypic difference between mice fed lard or those fed fish oil. Mice fed a lard diet have raised TLR activation in the systemic circulation, increased WAT inflammation and impaired insulin sensitivity as compared to mice fed fish oil. Mice which are deficient in Myeloid Differentiation factor 88 (MyD88) or TRIF are protected against lard induced WAT inflammation and metabolic perturbations and that saturated dietary lipids and gut microbiota interact to induce WAT inflammation. Additionally they showed CCL2 is required for microbiota induced macrophage recruitment to WAT in mice on a lard diet and that expression of CCL2 in adipocytes is induced by factors in the blood of lard fed CONV-R mice through a mechanism involving MyD88, TRIF and TLR4.
They further showed that type of dietary fat is a major driver, which influences both composition and diversity of gut microbiota. In mice fed fish oil Lactobacillus is known as a probiotic which has been linked to reduced inflammation and mucosal lesions scores in various models of inflammatory bowel disease and Akkermenia mucinphilia which has been shown to reduce fat mass gain and WAT macrophage infiltration and improve gut barrier function and glucose metabolism when administered in mice with Diet Induced Obesity (DIO) [19,20]. In contrast mice fed lard had increased levels of Bilophila. Earlier studies have shown that Bilophila increases in mice and humans after consumption of diets rich in saturated fats of animal origin and bilophila wadsworthia has been shown to exacerbate colitis in genetically susceptible models [21,22]. Hence to see if microbiota of fish oil fed mice could give protection against lard DIO and inflammation they transported microbiota from lard or fish oil fed mice into antibiotic treated mice that were then fed a lard diet for 3 weeks. Here Caesar et al., utilized antibiotic treated mice in contrast to Germ Free (GF) mice as it is known that GF mice have an underdeveloped immune system, which could potentially confound this analysis [23]. Mice receiving microbiota from lard fed donor showed increased adiposity and inflammation together with a significant increase in Lactobacillus as compared to mice that received microbiota from fish oil fed donor. Thus these data supported Lactobacillus in decreasing inflammation. But they found that the enrichment of Akkermansia co-occurred with partial protection against adiposity and inflammation in mice transplanted with fish oil microbiota and fed a lard diet highlighting Akkermansia as a potential mediator of the improved inflammation and metabolic phenotype of mice fed fish oil. This finding is in agreement with previous findings linking Akkermansia muciniphilia with protection against DIO [20,24]. Serum from mice fed lard had increased capacity to activate TLR4, which has been linked to WAT inflammation and metabolic perturbations [10,14,25]. Further they found that mice deficient in either of 2 TLR adaptor molecules MyD88 and TRIF were protected from lard induced WAT inflammation and insulin sensitivity. These findings are consistent with earlier studies showing decreased inflammation in mouse models lacking functional MyD88 [26-29]. One report has shown that MyD88 (Myeloid Differentiation factor 88) protects against glucose homeostasis perturbations and liver diseases during high fat diet [30]. Inconsistencies in report on the role of MyD88 is due to environmental factors at various animal facilities e.g., the presence of segmented filamentous bacteria which are enriched in MyD88-/- mice and have a major impact on host immunity differ between animal facilities [31-33]. Importantly the TRIF deficient mice in their study had the same body weight and adipocyte size as the wild type mice showing protection against WAT inflammation was not dependent on reduced adiposity. TLR signaling can be activated by both microbial and endogenous ligands and some investigators have suggested that saturated FA’s promote inflammation and IR in obesity through TLR4 [34]. The gut microbiota modulates host lipid metabolism [35]. Therefore protection against WAT inflammation in MyD88-/- and TRIF-/- mice fed lard might be due to reduced TLR signaling induced by ligands originating from the host or from the diet. Cesar et al., showed that serum levels of Lipopolysaccharides (LPS) were higher in mice fed lard as compared to mice fed fish oil which indicated that microbial factors are present in the periphery which may directly affect WAT inflammation. However they could not exclude the possibility of other factors like saturated lipids could also directly contribute to the inflammatory response by activating TLR signaling [36]. Further to determine the specific impact of the gut microbiota on lard induced WAT inflammation they compared the effect of lard and fish oil in CONV-R versus GF mice. GF mice were partly protected against lard induced WAT inflammation though the protection against obesity in GF mice was less than that observed in previous studies [9-12]. This is likely due to the reduced sucrose levels in the HFD‘s used in the present study. Sucrose levels have previously been shown to have a major impact on microbiota induced obesity [37]. Thus they used this fact as they could utilize weight matched mice to try and interfere whether the microbiota modulated WAT inflammation by weight dependent or independent mechanisms. They observed an adiposity independent link between the gut microbiota and WAT inflammation which may complicate microbially derived products as mediators of inflammation through TLR’s. Yet they also showed that GF mice fed lard had increased WAT inflammation compared to GF mice fed fish oil, indicating that microbiota independent mechanisms contribute to accumulation of immune cells in WAT. Previous studies have demonstrated that gut microbiota derived factors can induce inflammation and TNFα expression in WAT and they showed that serum from lard fed CONV-R mice compared with GF mice had an increased capacity to induce expression of Tumor Necrosis Factor Alpha (TNFα) in both adipocytes and macrophages [10]. CCL2 is the only chemokine which has been shown to mediate inflammation in a WAT specific knockout model and TLR ligand have been shown to induce secretion of CCL2 from 3T3L1 adipocytes [38,39]. Here they found that that CCL2 expression in primary adipocytes and WAT was induced by microbial factors in serum and required the presence of MyD88, TRIF and TLR4. Over expression of CCL2 in adipocytes has been shown to result in WAT inflammation and IR without obesity, and mice deficient in CCL2 or its receptor chemokine (C+C motif) receptor2 (CCR2) have reduced WAT inflammation and IR [40,41]. Also a recent study reported that CCL2 promotes local proliferation of macrophages in WAT [42]. By using the specific pharmacological CCL2 inhibitor MnOx E3 they demonstrated that CCL2 is essential for WAT macrophage accumulation in their model and therefore constitutes putative mediator of gut microbiota induced WAT inflammation [43-45]. In addition they found that GF mice fed lard or fish oil had similar expression levels of CCL2 in WAT, suggesting that microbiota independent WAT inflammation is not mediated through CCL2. Taken together their data showed that interaction between gut microbiota and dietary lipids induces WAT inflammation. They also identified putative mechanisms including role of cell signaling components and regulation of chemokine expression. Thus this study establishes the gut microbiota as an independent factor aggravating inflammation during DIO and therefore a suitable target for therapies against associated metabolic perturbations [18].
They further showed that type of dietary fat is a major driver, which influences both composition and diversity of gut microbiota. In mice fed fish oil Lactobacillus is known as a probiotic which has been linked to reduced inflammation and mucosal lesions scores in various models of inflammatory bowel disease and Akkermenia mucinphilia which has been shown to reduce fat mass gain and WAT macrophage infiltration and improve gut barrier function and glucose metabolism when administered in mice with Diet Induced Obesity (DIO) [19,20]. In contrast mice fed lard had increased levels of Bilophila. Earlier studies have shown that Bilophila increases in mice and humans after consumption of diets rich in saturated fats of animal origin and bilophila wadsworthia has been shown to exacerbate colitis in genetically susceptible models [21,22]. Hence to see if microbiota of fish oil fed mice could give protection against lard DIO and inflammation they transported microbiota from lard or fish oil fed mice into antibiotic treated mice that were then fed a lard diet for 3 weeks. Here Caesar et al., utilized antibiotic treated mice in contrast to Germ Free (GF) mice as it is known that GF mice have an underdeveloped immune system, which could potentially confound this analysis [23]. Mice receiving microbiota from lard fed donor showed increased adiposity and inflammation together with a significant increase in Lactobacillus as compared to mice that received microbiota from fish oil fed donor. Thus these data supported Lactobacillus in decreasing inflammation. But they found that the enrichment of Akkermansia co-occurred with partial protection against adiposity and inflammation in mice transplanted with fish oil microbiota and fed a lard diet highlighting Akkermansia as a potential mediator of the improved inflammation and metabolic phenotype of mice fed fish oil. This finding is in agreement with previous findings linking Akkermansia muciniphilia with protection against DIO [20,24]. Serum from mice fed lard had increased capacity to activate TLR4, which has been linked to WAT inflammation and metabolic perturbations [10,14,25]. Further they found that mice deficient in either of 2 TLR adaptor molecules MyD88 and TRIF were protected from lard induced WAT inflammation and insulin sensitivity. These findings are consistent with earlier studies showing decreased inflammation in mouse models lacking functional MyD88 [26-29]. One report has shown that MyD88 (Myeloid Differentiation factor 88) protects against glucose homeostasis perturbations and liver diseases during high fat diet [30]. Inconsistencies in report on the role of MyD88 is due to environmental factors at various animal facilities e.g., the presence of segmented filamentous bacteria which are enriched in MyD88-/- mice and have a major impact on host immunity differ between animal facilities [31-33]. Importantly the TRIF deficient mice in their study had the same body weight and adipocyte size as the wild type mice showing protection against WAT inflammation was not dependent on reduced adiposity. TLR signaling can be activated by both microbial and endogenous ligands and some investigators have suggested that saturated FA’s promote inflammation and IR in obesity through TLR4 [34]. The gut microbiota modulates host lipid metabolism [35]. Therefore protection against WAT inflammation in MyD88-/- and TRIF-/- mice fed lard might be due to reduced TLR signaling induced by ligands originating from the host or from the diet. Cesar et al., showed that serum levels of Lipopolysaccharides (LPS) were higher in mice fed lard as compared to mice fed fish oil which indicated that microbial factors are present in the periphery which may directly affect WAT inflammation. However they could not exclude the possibility of other factors like saturated lipids could also directly contribute to the inflammatory response by activating TLR signaling [36]. Further to determine the specific impact of the gut microbiota on lard induced WAT inflammation they compared the effect of lard and fish oil in CONV-R versus GF mice. GF mice were partly protected against lard induced WAT inflammation though the protection against obesity in GF mice was less than that observed in previous studies [9-12]. This is likely due to the reduced sucrose levels in the HFD‘s used in the present study. Sucrose levels have previously been shown to have a major impact on microbiota induced obesity [37]. Thus they used this fact as they could utilize weight matched mice to try and interfere whether the microbiota modulated WAT inflammation by weight dependent or independent mechanisms. They observed an adiposity independent link between the gut microbiota and WAT inflammation which may complicate microbially derived products as mediators of inflammation through TLR’s. Yet they also showed that GF mice fed lard had increased WAT inflammation compared to GF mice fed fish oil, indicating that microbiota independent mechanisms contribute to accumulation of immune cells in WAT. Previous studies have demonstrated that gut microbiota derived factors can induce inflammation and TNFα expression in WAT and they showed that serum from lard fed CONV-R mice compared with GF mice had an increased capacity to induce expression of Tumor Necrosis Factor Alpha (TNFα) in both adipocytes and macrophages [10]. CCL2 is the only chemokine which has been shown to mediate inflammation in a WAT specific knockout model and TLR ligand have been shown to induce secretion of CCL2 from 3T3L1 adipocytes [38,39]. Here they found that that CCL2 expression in primary adipocytes and WAT was induced by microbial factors in serum and required the presence of MyD88, TRIF and TLR4. Over expression of CCL2 in adipocytes has been shown to result in WAT inflammation and IR without obesity, and mice deficient in CCL2 or its receptor chemokine (C+C motif) receptor2 (CCR2) have reduced WAT inflammation and IR [40,41]. Also a recent study reported that CCL2 promotes local proliferation of macrophages in WAT [42]. By using the specific pharmacological CCL2 inhibitor MnOx E3 they demonstrated that CCL2 is essential for WAT macrophage accumulation in their model and therefore constitutes putative mediator of gut microbiota induced WAT inflammation [43-45]. In addition they found that GF mice fed lard or fish oil had similar expression levels of CCL2 in WAT, suggesting that microbiota independent WAT inflammation is not mediated through CCL2. Taken together their data showed that interaction between gut microbiota and dietary lipids induces WAT inflammation. They also identified putative mechanisms including role of cell signaling components and regulation of chemokine expression. Thus this study establishes the gut microbiota as an independent factor aggravating inflammation during DIO and therefore a suitable target for therapies against associated metabolic perturbations [18].
Role of gut immune system
Luck et al., showed that the gut immune system is altered during HFD feeding and is a functional regulator of obesity related IR which can be explored therapeutically. Obesity induces a chronic phenotypic proinflammatory shift in bowel lamina propria immune cell populations. Reducing the gut immune system using β7 integrin deficient mice decreased HFD induced IR. Treatment of wild type HFD C57BL/6 mice with the local gut anti-inflammatory 5-Aminosalicylic acid (5-ASA) reversed bowel inflammation and improves metabolic parameters. These beneficial effects are dependent on adaptive and gut immunity and are associated with reduced gut immunity and are associated with decreased gut permeability, endotoxaemia, decreased visceral adipose tissue inflammation and improved antigen specific tolerance to luminal antigens. Thus the mucosal immune system affects multiple pathways associated with systemic IR and represents a novel therapeutic target in this disease [46].
It is now accepted that both obesity and T2D are associated with low grade inflammation and that AT appears to be the first organ which is affected [47]. The development of inflammation and oxidative stress in AT leads to hepatic lipogenic expression and reduced liver fat export may also leads to obesity development [48,49].
It is now accepted that both obesity and T2D are associated with low grade inflammation and that AT appears to be the first organ which is affected [47]. The development of inflammation and oxidative stress in AT leads to hepatic lipogenic expression and reduced liver fat export may also leads to obesity development [48,49].
Role of Herbal Therapy
Many naturally occurring polyphenols have antioxidant and anti-inflammatory properties [50]. This could be achieved by modulating an inflammatory or oxidative signaling pathway [50-52]. Certain dietary phenols like curcumin, which is a low molecular weight polyphenols derived from herbal remedy and dietary spice turmeric, was found to prevent obesity and DM in mouse models [53]. Mechanistically curcumin may exert its beneficial effect via decreasing IR and leptin resistance attenuating inflammatory cytokine expression, accelerating fatty acid oxidation as well as increasing antioxidant enzyme expression [50]. Additionally curcumin could also function as an inhibitor of Histone Acetyltransferase p300 (HAT), a potential marker for cancer prevention and CVS complication [54,55]. The role of the canonical Wnt signaling pathway has recently received increasing attention. Activation of Wnt pathway increases cellular and nuclear β-catenin levels which represses adipogenesis while inhibition of Wnt signaling is required for PPARγ induction and preadipocyte differentiation [56]. A study showed that curcumin stimulates Wnt/β-cat signaling in 3T3L1 preadipocytes and hence suppresses adipogenic differentiation [57]. This gives a potential molecular mechanism for the effect of curicumin in attenuating obesity, although it is contradictory with other reports. First various studies have indicated that curcumin exerts its anticancer effects via repressing Wnt signaling [58,59]. Second Wnt activation in mature adipocytes was shown to induce IR [53].
In this study carried out by Shao et al., a HFD mouse model where development of obesity and insulin sensitivity was relatively slow due to the administration of 45% rather than 60% of calories from fat [8]. In this mouse model as well as in primary rat adipocytes they did not observe stimulation of curcumin on Wnt pathway components or Wnt target gene expression. But curcumin attenuated lipogenic gene expression in hepatocytes and blocked the effect of HFD on the inflammatory response in the AT associated with decreased weight/fat gain and the maintenance of normal glucose tolerance and insulin sensitivity. Thus they concluded that beneficial effects of curcumin during HFD consumption is mediated by attenuating lipogenic gene expression in the liver and the inflammatory response in AT, in the absence of Wnt signaling in mature adipocytes [60].
In this study carried out by Shao et al., a HFD mouse model where development of obesity and insulin sensitivity was relatively slow due to the administration of 45% rather than 60% of calories from fat [8]. In this mouse model as well as in primary rat adipocytes they did not observe stimulation of curcumin on Wnt pathway components or Wnt target gene expression. But curcumin attenuated lipogenic gene expression in hepatocytes and blocked the effect of HFD on the inflammatory response in the AT associated with decreased weight/fat gain and the maintenance of normal glucose tolerance and insulin sensitivity. Thus they concluded that beneficial effects of curcumin during HFD consumption is mediated by attenuating lipogenic gene expression in the liver and the inflammatory response in AT, in the absence of Wnt signaling in mature adipocytes [60].
Recapitulating the Control of Energy Homeostasis
There is a negative feedback system via which signals in the circulation like leptin send signals to important brain centers about body energy stores. Once change of body fat mass occurs there are corresponding changes of both energy intake and energy expenditure which favor return of body weight to preintervention levels. In response to weight loss due to calorie restriction, this model predicts that falling plasma levels of leptin and other adiposity negative signals e.g., insulin elicit brain responses which minimize further weight loss and promote eventual recovery of lost weight. Leptin signals mainly to modify activity of neuronal subsets in the arcuate nucleus of hypothalamus like the orexigenic agouti related peptide (AgRP/NPY) or the anorexigenic proopiomelanocortin (POMC/CART) neurons to decrease appetite and increase energy expenditure (Figure 1) [61]. Thus it allows lean people to be able to maintain stable body weight over many years, but fails to protect obese people from harmful effects of highly palatable calorie dense foods and sedentary lifestyles [62]. This differential susceptibility occurs not from leptin deficiency in obese people but due to acquired insensitivity to leptin action, which can be compared to central pathogenic role of insulin resistance in type 2 DM [63]. First time the group of Licio Velloso at the state university of Caminas in Brazil identified an inflammatory response in hypothalamus similar to that seen in peripheral tissues [64]. Rats fed a lard based High Fat Diet (HFD) increased mediobasal hypothalamic activation of the inflammatory signal intermediate c-Jun-n terminal Kinase (JNK) and Nuclear Factor kappa-B (NF?B) leads to production of proinflammatory cytokines (TNFα, IL1β and IL6) and impairment of insulin and leptin signaling.
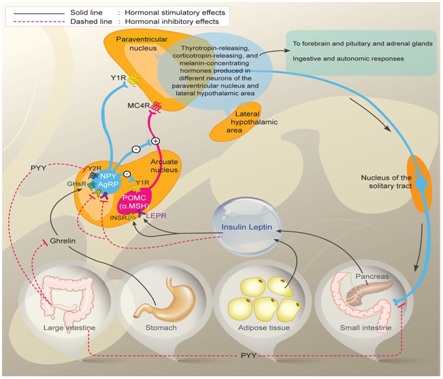
Figure 1: Control of Energy homeostasis.
Diagram depicting Orexigenic Agouti related peptide (AgRP)/Neuropeptide Y neurons (Agrp/NPY) and anorexigenic proopiomelanocirtin neurons (POMC/CART) in arcuate nucleus of hypothalamus and their regulation by circulating signals like leptin produced by adipocytes and insulin from beta cells of pancreas in energy homeostasis, besides role of ghrelin and Peptide YY(PYY) on orexegenic control acting through their receptors respectively. Also interconnection of arcuate nucleus with paraventricular nucleus and further with lateral hypothalamic areas is depicted and also over Nucleus Tract of Solitary nucleus (NTS) in hindbrain along with autonomic nervous system in regulation of energy homeostasis is depicted.
Hypothalamic Inflammation (HI)
Leptin resistance may be an acquired response or due to inherited response to leptin. Leptin resistance can potentially arise from a number of defects;
• Those affecting leptin receptor signal transduction
• At innumerable points downstream or in parallel to leptin mediated effects e.g., obesity secondary to Mc 4R mutation, the most common monogenic form of human obesity is characterized by leptin resistance, since leptin action on energy balance needs that it activate the melanocortin pathway [65]. The term leptin resistance is misused here because a blunted feeding response to leptin is present in practically all forms of obesity, except for that caused by leptin deficiency [65]. Hence it has become difficult to find the underlying mechanisms. Despite this problem the role of leptin resistance in obesity pathogenesis along with hypothalamic inflammation underlie this resistance. Following strong evidence that inflammatory signaling leads to obesity associated insulin resistance in peripheral tissues like liver, muscle and adipose tissue, this hypothesis was proposed [66]. Neuronal consequences of proinflammatory signaling is a disruption of intracellular signal transduction downstream of both insulin and leptin receptors via the insulin receptor substrate-phosphatidyl inositol 3 kinase pathway [67].
• Cellular inflammation can affect the leptin signaling through janus kinase-signal transducer and activator of transcription pathway [68,69]. This suggests that the mechanism may add not only to obesity associated leptin and insulin resistance (LR&IR) but also to the associated increase in the defended level of body fat stores. Still it is difficult to differentiate cause and effect and evidence of leptin resistance caused by inflammation independent of obesity is lacking through studies of ageing, injury and endotoxin induced leptin resistance which suggests this possibility [65,66,68].
Another mechanism by which HI is linked to IR and LR is via up regulation of Suppressor Of Cytokine Signaling 3 (SOCS3), which is a member of protein family originally characterized as negative feedback regulators of inflammation [68,70]. SOCS3 inhibits insulin and leptin signaling both by direct binding to their cognate receptors and targeting IRS proteins for proteasomal degradation [68,70]. HF feeding increases SOCS3 expression specifically within the ARC nucleus of hypothalamus coincident with the onset of LR selectively in this brain area [71]. The mechanism underlying increased SOCS3 expression during HFD is uncertain as it can be induced via either leptin-JAK/STAT or IKKβ/NF?B pathways.
Conversely, both SOCS3 haploin sufficiency and neuron specific SOCS3 deletion protect mice from DIO by increasing leptin sensitivity, while over expression of SOCS3 in POMC neurons (directly or by increasing STAT3 activation) results in hyperphagia and obesity on a chow diet [72-75]. Linking SOCS3 to inflammation, HFD resistant neuronal IKKβ knockout mice show greatly decreased SOCS3 expression in the hypothalamus, while misexpression of SOCS3 in the MBH abolishes protection from HFD induced obesity in AgRP neuron specific IKKβ knockout animals [76]. More experiments are needed to identify mechanisms underlying the hypothalamus specific increase of SOCS3 expression seen during HFD feeding and if HFD induced weight gain caused by augmenting HI needs functional SOCS3 signaling [71].
Like SOCS3, the protein tyrosine phosphatase (PTB)-1B is a signal terminal molecule which inhibits both leptin and insulin signaling. The mechanism underlying these effects involves its ability to dephosphorylate the insulin receptor JAK2 and more distal components of both pathways and available data suggest HF feeding increases PTB-1B expression in several tissues including the hypothalamus [77,78]. That this effect gets recapitulated by systemic TNFα administration suggests the functional interactions exist between inflammatory signaling and PTB-1B activation [77]. Pan-neuronal, POMC neuron specific PTB1B knockout mice are resistant to DIO due to enhanced hypothalamic leptin and insulin sensitivity, but how this response might be related to alter hypothalamic inflammatory signaling needs further studies [79]. Since both pan-neuronal PTB1B knockout mice and rats with hypothalamic PTB1B knockout show equivalent reductions of food intake whether fed chow or HFD, this protein may favor weight gain via mechanisms in addition to those involving hypothalamic inflammation [79,80] (Figure 2).
Thaler et al., proposes a hypothesis that a vicious cycle exists involving obesity and leptin resistance and inflammation. Diet induced increases of inflammation get a state of leptin resistance which promotes weight gain which in turn triggers further inflammation and leptin resistance ultimately leads to biological defense of an increased level of body functions. This perspective highlights the challenges inherent in determining the extent of which leptin resistance and inflammation are causes or consequences of weight gain, a challenge not possible to be met with use of methods which distinguish cellular response to diet from metabolic alterations induced by obesity itself [81].
• Those affecting leptin receptor signal transduction
• At innumerable points downstream or in parallel to leptin mediated effects e.g., obesity secondary to Mc 4R mutation, the most common monogenic form of human obesity is characterized by leptin resistance, since leptin action on energy balance needs that it activate the melanocortin pathway [65]. The term leptin resistance is misused here because a blunted feeding response to leptin is present in practically all forms of obesity, except for that caused by leptin deficiency [65]. Hence it has become difficult to find the underlying mechanisms. Despite this problem the role of leptin resistance in obesity pathogenesis along with hypothalamic inflammation underlie this resistance. Following strong evidence that inflammatory signaling leads to obesity associated insulin resistance in peripheral tissues like liver, muscle and adipose tissue, this hypothesis was proposed [66]. Neuronal consequences of proinflammatory signaling is a disruption of intracellular signal transduction downstream of both insulin and leptin receptors via the insulin receptor substrate-phosphatidyl inositol 3 kinase pathway [67].
• Cellular inflammation can affect the leptin signaling through janus kinase-signal transducer and activator of transcription pathway [68,69]. This suggests that the mechanism may add not only to obesity associated leptin and insulin resistance (LR&IR) but also to the associated increase in the defended level of body fat stores. Still it is difficult to differentiate cause and effect and evidence of leptin resistance caused by inflammation independent of obesity is lacking through studies of ageing, injury and endotoxin induced leptin resistance which suggests this possibility [65,66,68].
Another mechanism by which HI is linked to IR and LR is via up regulation of Suppressor Of Cytokine Signaling 3 (SOCS3), which is a member of protein family originally characterized as negative feedback regulators of inflammation [68,70]. SOCS3 inhibits insulin and leptin signaling both by direct binding to their cognate receptors and targeting IRS proteins for proteasomal degradation [68,70]. HF feeding increases SOCS3 expression specifically within the ARC nucleus of hypothalamus coincident with the onset of LR selectively in this brain area [71]. The mechanism underlying increased SOCS3 expression during HFD is uncertain as it can be induced via either leptin-JAK/STAT or IKKβ/NF?B pathways.
Conversely, both SOCS3 haploin sufficiency and neuron specific SOCS3 deletion protect mice from DIO by increasing leptin sensitivity, while over expression of SOCS3 in POMC neurons (directly or by increasing STAT3 activation) results in hyperphagia and obesity on a chow diet [72-75]. Linking SOCS3 to inflammation, HFD resistant neuronal IKKβ knockout mice show greatly decreased SOCS3 expression in the hypothalamus, while misexpression of SOCS3 in the MBH abolishes protection from HFD induced obesity in AgRP neuron specific IKKβ knockout animals [76]. More experiments are needed to identify mechanisms underlying the hypothalamus specific increase of SOCS3 expression seen during HFD feeding and if HFD induced weight gain caused by augmenting HI needs functional SOCS3 signaling [71].
Like SOCS3, the protein tyrosine phosphatase (PTB)-1B is a signal terminal molecule which inhibits both leptin and insulin signaling. The mechanism underlying these effects involves its ability to dephosphorylate the insulin receptor JAK2 and more distal components of both pathways and available data suggest HF feeding increases PTB-1B expression in several tissues including the hypothalamus [77,78]. That this effect gets recapitulated by systemic TNFα administration suggests the functional interactions exist between inflammatory signaling and PTB-1B activation [77]. Pan-neuronal, POMC neuron specific PTB1B knockout mice are resistant to DIO due to enhanced hypothalamic leptin and insulin sensitivity, but how this response might be related to alter hypothalamic inflammatory signaling needs further studies [79]. Since both pan-neuronal PTB1B knockout mice and rats with hypothalamic PTB1B knockout show equivalent reductions of food intake whether fed chow or HFD, this protein may favor weight gain via mechanisms in addition to those involving hypothalamic inflammation [79,80] (Figure 2).
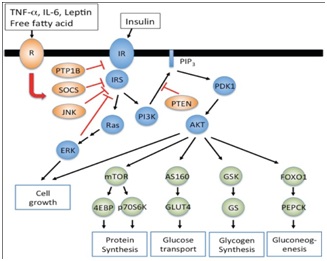
Figure 2: Demonstrating actions of leptin, insulin, free fatty acids on their receptors and downstream effects on SOCS3 and PTB1B.
Figure depicting how leptin, cytokines free fatty acids act on their receptors as does insulin and downstream effects of common signaling pathways like SOCS3, Protein tyrosine phosphatase 1 B effect glucose transport glucose sensing and various aspects of energy homeostasis.
Thaler et al., proposes a hypothesis that a vicious cycle exists involving obesity and leptin resistance and inflammation. Diet induced increases of inflammation get a state of leptin resistance which promotes weight gain which in turn triggers further inflammation and leptin resistance ultimately leads to biological defense of an increased level of body functions. This perspective highlights the challenges inherent in determining the extent of which leptin resistance and inflammation are causes or consequences of weight gain, a challenge not possible to be met with use of methods which distinguish cellular response to diet from metabolic alterations induced by obesity itself [81].
HYPOTHALAMUS AND INSULIN RESISTANCE (IR)
IR gets initiated at the hypothalamus which tries to balance energy intake with energy expenditure in a way to prevent over deposition of stored energy [82]. Satiety signals from the gut are matched by adiposity (mainly leptin) and blood (primarily insulin) hormonal signals to control food intake [67,82]. Both saturated fats like palmitic acid or abundant calories can cause inflammation in hypothalamus => resistance to the satiety signaling of both insulin and leptin [83-86]. This satiety is repressed and there is increased appetite. Also there are GPR120 binding proteins which are specific for long chain omega 3 fatty acids like Eicosa Pentaenoic Acid (EPA) and Docosa Hexaenoic Acid (DHA) [86]. Hence the presence of adequate levels of these omega 3 fatty acids in the diet can reduce inflammation within the hypothalamus [86]. ICV injection of omega-3 fatty acids into obese rats reduces insulin resistance [86-88]. Injecting anti-TLR4 and anti TNFα antibodies ICV also reduce IR [88]. HFD especially those rich in saturated fats are the usual methods to cause DIO in animal models. Increased inflammation appears within 24hrs after beginning a HFD as indicated by increase in JNK and IKK proteins as well as increased expression of TLR-4 receptors and detection of ER stress [61]. IKK induces inflammation, while activation of NF?B inhibits the normal hormonal signaling of leptin and insulin essential to create satiety. JNK activation is often preceded by the increase in ER stress [89]. This sets up a vicious cycle of increase in hunger and ultimately leads to accumulation of excess calories as stored fat in the AT. Inflammation in hypothalamus precedes any weight gain in AT [86]. This also explains why significant calorie restriction can decrease IR before any major loss in increased body fat in the AT. Increased nutrient intake especially saturated fat can also indirectly cause inflammation in the hypothalamus by the activation of TLR4 in the microglia in the brain which ultimately causes damage to neurons in the hypothalamus [84]. With extended use of HFD there is a decrease in number of neurons which are responsible for generating satiety signals in hypothalamus namely POMC neurons [90]. Also HFD’s are associated with increased production of palmitic acid enriched ceramides in the hypothalamus [2]. This would provide another link to increased IR and Leptin resistance which leads to increased hunger as satiety depends on functioning insulin pathways in the hypothalamic neurons [91]. Although GPR 120 receptors in the hypothalamus are the ones which are activated by omega-3 fatty acids which reduce inflammation, there are other fatty acid nutrient sensors in the hypothalamus which can be activated to increase inflammation [91,92]. Any rise in FFA levels in the blood can be sensed by the CD36/FATP1 transporter at the same surface of BBB. If these FA’s are rich in palmitic acid the primary product of de novo lipid production in the liver caused by excessive dietary glucose, then the HPA axis is activated to release more cortisol there by increasing IR [93]. Conversely if the FA being sensed is primarily oleic acid there will be a reduction in NPY (a powerful appetite inducing hormone) expression in the hypothalamus which promotes satiety [94].
Also there is interaction of the hypothalamus with the liver via signaling through the vagus nerve [95]. This explains why inhibiting TNFα or TLR4 signaling in the hypothalamus also decreases glucose production in the liver. This implies that the central regulation of appetite control by hypothalamus is a very complex interaction of the levels of inflammation and nutrient intake generated by the diet and sensing of those levels by the hypothalamus.
Also there is interaction of the hypothalamus with the liver via signaling through the vagus nerve [95]. This explains why inhibiting TNFα or TLR4 signaling in the hypothalamus also decreases glucose production in the liver. This implies that the central regulation of appetite control by hypothalamus is a very complex interaction of the levels of inflammation and nutrient intake generated by the diet and sensing of those levels by the hypothalamus.
HYPOTHALAMIC INFLAMMATION (HI) AND OBESITY
Various human genome wide association studies have found loci near or within various neuronal genes which affect BMI which suggest that changes in central control of metabolism may play an important role in genetic obesity risk [96]. Both lipid infusion and HFD are associated with inflammatory signaling pathways which results in increased food intake and nutrient storage [67].
With DIO, metabolites like diacyl glycerol and ceramide accumulate in hypothalamus causing leptin and insulin resistance in CNS [97,98]. Part of this effect is mediated by saturated FA’s which activate neuronal JNK & NF?B signaling pathway which direct effects in leptin and insulin signaling. Disruption of signaling through TLR4, MyD88, IKKβ/NF?B/ER stress pathway in neurons protects mice from DIO and its downstream metabolic effects [75,99,100].
Thus brain inflammation has various influences on the peripheral tissue function. HI can affect insulin release from β cells as well as have an impact on peripheral insulin action and accentuate hypertension independent of obesity [101-103]. So many of these are a result of SNS signals which by themselves can also induce inflammatory changes in AT inflammation responses to neuronal injury [104]. What is still not clear is how inflammatory signals in the brain develop responses that create negative energy balance (anorexia) while simultaneously on the other hand can result in positive energy balance (weight gain) [105].
The marked interplay between HI and obesity suggest additional targets for anti-inflammatory treatment in obesity. That anti-inflammatory pathways might help to counterbalance CNS inflammation events and improve leptin sensitivity is what one infers from this observation. One that IL6 and IL10 are involved in exercise induced suppression of hyperphagia and suppresses IKKβ/NF?B and ER stress in the brain [106]. Second that the IKKβ/NF?B inhibitor sodium salicylate can also prevent ceramide getting deposited in hypothalamus with lipid infusion [98].
With DIO, metabolites like diacyl glycerol and ceramide accumulate in hypothalamus causing leptin and insulin resistance in CNS [97,98]. Part of this effect is mediated by saturated FA’s which activate neuronal JNK & NF?B signaling pathway which direct effects in leptin and insulin signaling. Disruption of signaling through TLR4, MyD88, IKKβ/NF?B/ER stress pathway in neurons protects mice from DIO and its downstream metabolic effects [75,99,100].
Thus brain inflammation has various influences on the peripheral tissue function. HI can affect insulin release from β cells as well as have an impact on peripheral insulin action and accentuate hypertension independent of obesity [101-103]. So many of these are a result of SNS signals which by themselves can also induce inflammatory changes in AT inflammation responses to neuronal injury [104]. What is still not clear is how inflammatory signals in the brain develop responses that create negative energy balance (anorexia) while simultaneously on the other hand can result in positive energy balance (weight gain) [105].
The marked interplay between HI and obesity suggest additional targets for anti-inflammatory treatment in obesity. That anti-inflammatory pathways might help to counterbalance CNS inflammation events and improve leptin sensitivity is what one infers from this observation. One that IL6 and IL10 are involved in exercise induced suppression of hyperphagia and suppresses IKKβ/NF?B and ER stress in the brain [106]. Second that the IKKβ/NF?B inhibitor sodium salicylate can also prevent ceramide getting deposited in hypothalamus with lipid infusion [98].
CERAMIDES AND INTRACELLULAR LIPIDS IN INFLAMMATION/METABOLISM
The downstream effects of TLR4 activation are not limited to the activation of NF?B. An important link between metabolism and inflammation may be closely linked to the balance between intracellular lipid species like ceramides and sphingolipids [107,108]. Inhibition of ceramide production blocks the ability of saturated FA’s to induce insulin resistance [109]. Ceramide synthesis by LPS and saturated FA’s is dependent upon TLR4 in many metabolic tissues include the hypothalamus and muscle where ceramide production in metabolic tissue is dependent on IKKβ as salicylates decrease ceramide levels in the liver, muscle and hypothalamus.
HYPOTHALAMIC GLIOSES AND NEURON INJURY
At the cellular level exposure of neurons to nutrient excess presents a significant stress which not only engaged adaptive mechanisms like autophagy and ER stress which limits neuronal damage, but also involves neighboring cell populations. Greater than 50% of the cellular components of the brain is non-neuronal, including glial, vascular and periventricular constituents [110]. Astrocytes and microglia are the most abundant of these specialized cell types and in addition involved in clearing degenerating cells. By amplifying inflammatory signals in the hypothalamus analogously to the recruitment and activation of proinflammatory immune cells in adipose and other peripheral tissues, both cell types have the potential to impair neuronal function in ways that favor leptin resistance and associated weight gain [111,112]. Lending support to this argument, TLR4 a putative mediator of saturated fatty acid induced inflammatory signaling, is abundantly expressed in microglia and acute inactivation of proinflammatory immune cells in adipose tissue and other peripheral tissues [85,112]. Both cell types have the potential to impair neuronal function in ways that favor leptin resistance and associated weight gain. Lending support to this argument, TLR4 a putative mediator of saturated fatty acid induced inflammatory signaling, is abundantly expressed by microglia [113]. Additionally reactive astrocytes and microglia accumulate in the hypothalamus during long term HFD consumption at times when hypothalamic inflammation is clearly evident [113,114]. In contrast, HFD fed mice subjected to involuntary exercise regimen exhibit modest improvements in glucose tolerance along with reduced hypothalamic micrtoglial activation, suggesting a link between microglial phenotype and obesity associated metabolic improvements [115]. Lastly, in a nonhuman primate model, microglial activation along with increased expression of inflammatory pathway genes was observed in fetuses from HFD fed mothers raising the possibility that hypothalamic microglia contribute to the effect of intrauterine programming to influence adult phenotype [116].
A more complex picture suggested by other studies regarding role of microglia and astrocytes in limiting the deleterious hypothalamic effects as a result of HFD consumption. Mice with moderately increased production of IL6 from astrocytes were protected from DIO, rather than being more susceptible [117]. Also adult rats overfed during the neonatal period manifest hypothalamic microglia activation (as evidenced by major histocompatibility complex class II expression) without local inflammation and hypothalamic microglia from mice fed an HFD accumulate the general anti-inflammatory molecule IgG [118,119]. Further work of Thaler et al., showed that effects of short term HFD feeding on hypothalamic inflammation and reactive glioses are separable from one another [113]. Although when both processes were seen within the first week of HFD consumption, hypothalamic inflammation subsided over the next 2 weeks despite accumulation of enlarged microglia in the ARC which continue unabated.
Despite definite answers are not there these results are consistent with the model in which glioses develops initially as a neuro protective response to cope with neuronal stress induced by HFD feeding. In this scenario, glial responses initially constrain hypothalamic inflammatory signaling. With prolonged exposure to an HFD, however astrocytes and microglia may eventually convert to a more proinflammatory, neurotoxic phenotype (Figure 3).
A more complex picture suggested by other studies regarding role of microglia and astrocytes in limiting the deleterious hypothalamic effects as a result of HFD consumption. Mice with moderately increased production of IL6 from astrocytes were protected from DIO, rather than being more susceptible [117]. Also adult rats overfed during the neonatal period manifest hypothalamic microglia activation (as evidenced by major histocompatibility complex class II expression) without local inflammation and hypothalamic microglia from mice fed an HFD accumulate the general anti-inflammatory molecule IgG [118,119]. Further work of Thaler et al., showed that effects of short term HFD feeding on hypothalamic inflammation and reactive glioses are separable from one another [113]. Although when both processes were seen within the first week of HFD consumption, hypothalamic inflammation subsided over the next 2 weeks despite accumulation of enlarged microglia in the ARC which continue unabated.
Despite definite answers are not there these results are consistent with the model in which glioses develops initially as a neuro protective response to cope with neuronal stress induced by HFD feeding. In this scenario, glial responses initially constrain hypothalamic inflammatory signaling. With prolonged exposure to an HFD, however astrocytes and microglia may eventually convert to a more proinflammatory, neurotoxic phenotype (Figure 3).
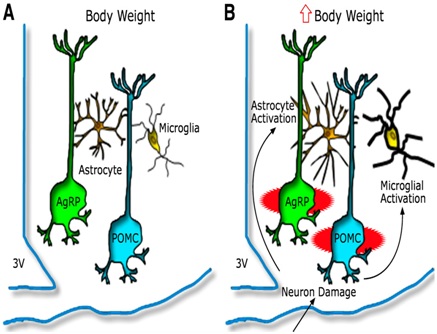
Figure 3: Hypothalamic inflammation and glioses leading to injury to arcuate neuronal subsets [81].
Model depicting the hypothalamic response to an HFD in animals predisposed to DIO A) AgRP and POMC neurons are integral components of energy balance neurocircuitry located in the ARC nucleus, situated adjacent to third cerebral ventricle (3V) along the floor of the hypothalamus. Activity of these neurons is sensitive to input from circulating hormones (e.g leptin and insulin) and nutrients and it plays an important role to establish the defended level of body weight. B) Recent evidence suggests that during HFD feeding these neurons may be injured via unknown mechanisms, and that this injury triggers activation of local glial cell populations (astrocytes and microglia). This neuron injury and reactive glioses can in turn impair homeostatic control of body fat stores, leading to increased body weight.
A growing literature in rodent models suggests that obesity is associated with inflammation of an injury to hypothalamic areas critical to the control of energy balance and glucose imbalance [113,119-122]. Histologically this response is characterized by gliosis, the proliferation and activation of glial cells induced by response to CNS injury. Microscopically gliosis means infiltration of microglia and astrocytes and astrocytic proliferation including increased density of astrocyte processes on the cell bodies on neurons.
Feeding rodents a HFD triggers inflammation and gliosis in the arcuate nucleus located in the Mediobasal Hypothalamus (MBH), even before obesity occurs and eventually reduces proopiomelanocortin cell number [113,121]. Such changes are associated with both obesity and impaired glucose homeostasis in rodents and they offer an explanation for obesity associated resistance of hypothalamic neurons in humoral signals like leptin and insulin [122-126]. Even though evidence exists from animal studies, significance of this hypothalamic glioses in humans had largely been unknown [118] (Figure 3).
A core concept of current research is that glioses can be detected in humans using MRI by assessing for increased signal brightness on a T2 weighted image [127-129]. Clinically the visual identification of increased T2 signal intensity is used to detect CNS insults like stroke and multiple sclerosis but quantitative techniques can detect more subtle alterations in CNS tissue characteristics [120, 127-129]. One prior retrospective study on humans utilized clinical MRI examination and found a positive association between BMI and ratio on T2 signal in the MBH as compared to the amygdale [113]. Thus Schur et al., in recent studies utilized a quantitative MRI technique to measure T2 relaxation time in the MBH, employing a dedicated sequence not typically utilized in clinical imaging protocols. Using a similar sequence they found longer MBH T2 relaxation times in DIO mice compared to chow fed controls [120,130]. Thus using 2 separate studies they sought evidence for MBH gliosis in human studies. In study 1 an in vivo MRI study it was hypothesized that MBH gliosis when present would be associated with obesity and insulin resistance. In study 2, a postmortem study of human brain tissue hypothesized that T2 relaxation time would be related to immunohistochemical measures of astrocytes in the MBH.
Schur et al., examined 67 patients who underwent a fasting blood draw and MRI, Cases with radiologic evidence of MBH gliosis [n=22] were identified as the upper tertile of left MBH T2 relaxation time and were compared to controls [n=23] from the lowest tertile. Besides a separate postmortem study brain slices [n =10] through the MBH was imaged by MRI and stained for Glial Fibrillary Acidic Protein (GFAP). In all participants longer T2 relaxation time in the left MBH was associated with higher BMI (P=0.01). As compared to control, cases had longer T2 relaxation times in the right MBH (P<0.05) as well as higher BMI (P<0.05), fasting insulin concentrations (p<0.01) and HOMA IR values (p<0.01) adjusted for sex and age. Elevations in insulin HOMA IR were also independent of BMI. In the postmortem study GFAP staining intensity was positively associated with MBH T2 relaxation time (p<0.05) validating an MRI based method for the detection of MBH gliosis in humans. Hence they concluded that these findings links hypothalamic gliosis to insulin resistance in humans and suggests that the link is independent of the level of adiposity [131].
ASTROCYTES AND HYPOTHALAMIC GLIOSES
Data from recent studies suggest that neuronal inflammation may be a downstream event during DIO, with recruitment and activation of hypothalamic glial cells being a more proximal response to HFD exposure [113,120,130-133]. This gliosis process involves accumulation and multiplication of activated microglia and astrocytes in the region of MBH [113,120,130,132,134]. Various studies have implicated microglia in the development of diet induced inflammatory signals along with metabolic dysfunction, but a similar role for astrocytes is not clear [123,135]. Buckman demonstrated modest role of astrocytic inflammation to caloric intake on the first day of HFD feeding but there was no analysis of susceptibility to DIO [136]. There are abundant astrocytes throughout the CNS and involved in many fundamental processes like synaptic transmission, neurovascular coupling and blood brain barrier maintenance [137]. Additionally astrocytes participate in CNS immune responses when they take an activated phenotype having raised GFAP expression and release of proinflammatory cytokines which can enhance neurotoxicity and neurodegenerative disease progression [137-139]. Hence astrocytes have the basic requirement to affect energy homeostasis regulation in health and disease. MBH astrocytes modulate feeding behavior on pharmacological activation and show dynamic responses to circulating signals of nutrient availability like insulin and leptin [140-145]. Also MBH astrocytes become activated with obesity and HFD feeding in rodents and humans, which raises the possibility that astrocyte inflammation disrupts hypothalamic regulation of energy balance and promotes DIO [113,131].
The group of Thaler et al., developed a mouse model with an inducible astrocytic specific deletion of IKKβ with the use of tamoxifen. With this approach they showed that decreasing the astrocytic signaling protects mice from HFD induced hypothalamic inflammation and decreases susceptibility to DIO and glucose tolerance. The results highlight the importance of non neuronal cells in obesity pathogenesis and suggest the possibility of newer target for therapy [146].
The group of Thaler et al., developed a mouse model with an inducible astrocytic specific deletion of IKKβ with the use of tamoxifen. With this approach they showed that decreasing the astrocytic signaling protects mice from HFD induced hypothalamic inflammation and decreases susceptibility to DIO and glucose tolerance. The results highlight the importance of non neuronal cells in obesity pathogenesis and suggest the possibility of newer target for therapy [146].
CONCLUSION
Thus in first part we have highlighted how gut microbiota contribute to phenotypic differences in mice fed high fat diet (lard or fish oil) with fish oil having not only differences with fermicutes and bacteroides in high fat diet but also more evidence of Lactobacillus & Akkermansia muciniphilia. Mice lacking MyD88 or TRIF are protected against WAT inflammation and microbial derived factor induce CCL2 in adipocytes TLR4, MyD88 and TRIF and this microbial induced CCL2 increases macrophage accumulation in WAT.
Further in both rodent models and humans now there is evidence that hypothalamic inflammation precedes the development of obesity, there is possibility that neuronal damage occurs with directly Ag RP and POMC neurons getting damaged by circulating HFD. Evidence is there that this neuronal injury triggers activation of local glial cells populations. This neuronal injury and reactive gliosis in turn may impair homeostatic control of body fat stores leads to increased body weight. Thus pharmacological targeting therapies at this level needs to be done to protect against obesity.
Further in both rodent models and humans now there is evidence that hypothalamic inflammation precedes the development of obesity, there is possibility that neuronal damage occurs with directly Ag RP and POMC neurons getting damaged by circulating HFD. Evidence is there that this neuronal injury triggers activation of local glial cells populations. This neuronal injury and reactive gliosis in turn may impair homeostatic control of body fat stores leads to increased body weight. Thus pharmacological targeting therapies at this level needs to be done to protect against obesity.
REFERENCES
- Kaur KK, Allahabadia G, Singh M (2017) A Review of Nutrient Metabolism in Obesity with Special Emphasis on Fatty Acid Metabolism. BAOJ Food Sci &Tec 1: 001.
- Kaur KK, Allahabadia G, Singh M (2016) An update on a etiopathogenesis and management of obesity. Obes Control Ther 3: 1-17.
- Kennedy A, Martinez K, Chuang CC, LaPoint K, Mcintosh M (2009) Saturated fatty acid-mediated inflammation and insulin resistance in adipose tissue: mechanisms of action and implications. J Nutr 139: 1-4.
- Buckley JD, Howe PR (2009) Anti-obesity effects of long-chain omega-3 polyunsaturated fatty acids. Obes Rev 10: 648-659.
- Calder PC (2006) n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am J Clin Nutr 83: 1505-1519.
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, et al. (2010) GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142: 687-698.
- Scott KP, Gratz SW, Sheridan PO, Flint HJ, Duncan SH (2013) The influence of diet on the gut microbiota. Pharmacol Res 69: 52-60.
- Tremarolli V, Backhead F (2012) Functional interactions between the gut microbiota and host metabolism. Nature 489: 242-249.
- Backhead F, Manchester JK, Samekovich CF, Gordon JI (2007) Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci USA 104: 979-984.
- Caesar R, Reigstad CS, Bachead HK, Reinhardt C, Ketonen M, et al. (2012) Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 61: 1701-1707.
- Ding S, Chi MM, Scull BP, Rigby R, Schwerbrock NMJ, et al. (2010) High-Fat Diet: Bacteria Interactions Promote Intestinal Inflammation Which Precedes and Correlates with Obesity and Insulin Resistance in Mouse. PLOS ONE 5: 12191.
- Rabot S, Membrez M, Brunea A, Gerard P, Harach T, et al. (2010) Germfree C57BL/6J mice are resistant to high fat diet induced insulin resistance and have cholesterol metabolism. FASEB J 24: 4948-4959.
- Caesar R, Fåk F, Bäckhed F (2010) Effects of gut microbiota on obesity and atherosclerosis via modulation of inflammation and lipid metabolism. J I nter Med 268: 320-328.
- Cani PD, Amar J, Igelesias MA, Poggi M, Knauf C, et al. (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56: 1761-1772.
- Henao-Mejia J, Elinav E, Jin C, Hao L, Mehao WZ, et al. (2012) Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. 482: 179-185.
- Jin C, Flavell RA (2013) Innate sensors of pathogen and stress: linking inflammation to obesity. J Allergy Clin Immunol 132: 287-294.
- Yu L, Wang L, Chen SJ (2010) Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med 14: 2592-2603.
- Caesar R, Tremorelli V, Datchary PK, Cani PD, Backhed F (2015) Crosstalk between Gut Microbiota and Dietary Lipids Aggravates WAT Inflammation through TLR Signaling. Cell Metab 22: 658-668.
- Guarner F, Perdigon G, Corthier G, Salminen S, Koletzko B, et al. (2005) Should yoghurt cultures be considered probiotic? Br J Nutr 93 : 783-786.
- Everard A, Belzer C, Geurts L, Quwerkerk JP, Druart C, et al. (2013) Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc Natl Acad Sci USA 110: 9066-9071.
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, et al. (2014) Diet rapidly and reproducibly alters the human gut microbiome. Nature 505: 559-563.
- Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach M, et al. (2012 ) Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature 487: 104-108.
- Hooper LV, Littman DR, Macpherson AG (2012) Interactions between the microbiota and the immune system. Science 336: 1268-1273.
- Shin NR, Lee JC, Lee HY, Kim MS, Whon TW, et al. (2014) An increase in the Akkermansiapopulation induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 63: 727-735.
- Creely SJ, McTernan PG, Kusminski CM, Fisher M, DaSilva NF, et al. (2007) Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab 292: 740-747.
- Bjorbacks H, Kunjathoor VV, Moore KJ, Koehn S, Ordija CM, et al. (2004) Reduced atherosclerosis in MyD88-null mice links elevated serum cholesterol levels to activation of innate immunity signaling pathways. Nat Med 10: 416-421.
- Everard A, Geurts IL, Caesar R, VanHul M, Matamoros S, et al. (2014) Intestinal epithelial MyD88 is a sensor switching host metabolism towards obesity according to nutritional status. Nat Commun 5: 5648.
- Michelsen KS, Wong MH, Shah PK, Zhang W, Yano J, et al. (2004) Lack of Toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc Natl Acad Sci USA 101: 10679-10684.
- Richards MR, Black AS, Bonnett DJ, Barish GD, Woo CW, et al. (2013) The LPS2 mutation in TRIF is atheroprotective in hyperlipidemic low density lipoprotein receptor knockout mice. Innate Immun 19: 20-29.
- Hosoi T, Yokoyama S, Matsuo S, Akira S, Ozawa K (2010) Myeloid Differentiation Factor 88 (MyD88)-Deficiency Increases Risk of Diabetes in Mice. PLoS One: 12537.
- Larson E, Tremarolli V, Lee YS, Koren O, Nookaew I, et al. (2012) Analysis of gut microbial regulation of host gene expression along the length of the gut and regulation of gut microbial ecology through MyD88. Gut 61: 1124-1131.
- Ivanov LL, Atarashi K, Manel N, Brodie EL, Shima T, et al. (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485-498.
- Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, et al. (2011) Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci USA 108: 11548-11553.
- Shi H, Kokoeva MV, Inouye K, Tzameli L, Yin H, et al. (2006) TLR4 links innate immunity and fatty acid induced insulin resistance. J Clin Invest 116: 3015-3025.
- Velagapudi VR, Hezaveh R, Reigstad CS, Gopalacharyulu P, Yetukuri L, et al. (2010) The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res 51: 1101-1112.
- Huang S, Rutkowsky JM, Snodgrass RG, Ono Moore KD, Schneider DA, et al. (2012) Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J Lipid Res 53 2002-2013.
- Fleissner CK, Hubel N, Abd-El Bary MM, Loh G, Klaus S, et al. (2010). Absence of intestinal microbiota does not protect mice from diet-induced obesity. Br J Nutr 104: 919-929.
- Lee BC, Lee J (2014) Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. J Biochym Biophys Acta 1842: 446-462.
- Kopp A, Buechler C, Numeier M, Weigert C, Scholmerich J, et al. (2009) Inn ate immu activnity and adipocyte function :ligand specific activation of multiple toll like receptors modulate cytokine ,adipokine, andchemokine secretion in adipocytes.Obesity(Silver Springs) 17: 648-656.
- Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, et al. (2006) Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 281: 26602-26614.
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, et al. (2006) CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116: 115-124.
- Amano SU, Cohen JL, Vangala P, Tencernova M, Nicoloro SM, et al. (2014) Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab 19: 162-171.
- Kulkarni O, Pawar RD, Purschke W, Eulberg D, Selve N, et al. (2007) Spiegelmer inhibition of CCL2/MCP-1 ameliorates lupus nephritis in MRL-(Fas)lpr mice. Am Soc Nephrol 18: 2350-2358.
- Kulkarni O, Eulberg D, Selve N, Zollner S, Allam R, et al. (2009) Anti-Ccl2 Spiegelmer Permits 75% Dose Reduction of Cyclophosphamide to Control Diffuse Proliferative Lupus Nephritis and Pneumonitis in MRL-Fas(lpr) Mice. J Pharmacol Exp Ther 328: 371-377.
- Wlotzka B, Leva S, Eschgfäller B,, Burmeister J, Kleinjung F, et al. (2002) In vivo properties of an anti-GnRH Spiegelmer: an example of an oligonucleotide-based therapeutic substance class. Proc Natl Acad Sci USA 99: 8898-8902.
- Luck H, Tsai S, Chung J, Clement Caesar X, Ghzarian M, et al. (2015) Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab 21: 527-542.
- Hotamisligil GS (2006) Inflammation and metabolic disorders. Nature 444: 860-867.
- Dentin R, Beuhamed F, Hainault I, Fauveau V, Foufelle F, et al. (2006) Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes 55: 2159-2170.
- Xu F, Gao Z, Zhang J, Rivera CA, Yin J, et al. (2010) Lack of SIRT1 (Mammalian Sirtuin 1) activity leads to liver steatosis in the SIRT1+/- mice: a role of lipid mobilization and inflammation. Endocrinology 151: 2504-2514.
- Allapat I, Assad AB (2010) Curcumin and obesity: evidence and mechanisms. Nat Rev 68: 729-738.
- Yu Z, Shao W, Chiang Y, Foltz W, Zhang Z, et al. (2010) Oltipraz upregulates the nuclear respiratory factor 2 alpha subunit (NRF2) antioxidant system and prevents insulin resistance and obesity induced by a high-fat diet in C57BL/6J mice. Diabetologia 54: 922-934.
- Breswill S, Munoz M, Fischer A, Plickert R, Haag LM, et al. (2010) Anti inflammatory effects of resveratrol, curcumin and simvastatin in acute small intestinal inflammation. PLoS One 5: 15099.
- Weisberg SP, Leibel R, Tortoriello DV (2008) Dietary curcumin significantly improves obesity-associated inflammation and diabetes in mouse models of diabesity. Endocrinology 149: 3549-3558.
- MorimotoT, Sunagawa Y, Kawamura T, Takaya T, Wada H et al. (2008) The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest 118: 868-878.
- Barnes PJ (2009) Role of HDAC2 in the pathophysiology of COPD. Annu Rev Physiol 71: 451-464.
- Krishnan V, Byant HU, Macdougald OA (2006) Regulation of bone mass by Wnt signaling. J Clin Invest 116: 1202-1209.
- Ahn J, Lee H, Kim S, Ha T (2010) Curcumin-induced suppression of adipogenic differentiation is accompanied by activation of Wnt/beta-catenin signaling. Am J Physiol Cell Physiol 298: 1510- 1516.
- Mukhopadyay A, Bannerjee S, Stafford IJ, Xia C, Liu M, et al. (2002) Curcumin-induced suppression of cell proliferation correlates with down-regulation of cyclin D1 expression and CDK4-mediated retinoblastoma protein phosphorylation. Oncogene 21:8852-8861.
- Jaiswal AS, Marlow BP, Gupta N, Narayan S (2002) Beta-catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene 21: 8414-8427.
- Shao W, Yu Z, Chiang Y, Yang Y, Chai T, et al. (2012) Curcumin prevents high fat diet induced insulin resistance and obesity via attenuating lipogenesis in liver and inflammatory pathway in adipocytes. PLoS One 7: 28784.
- Morton GJ, Cummings DE, Baskin DG, Barsch GS, Shwartz MW (2006) Central nervous system control of food intake and body weight. Nature 443:289-95.
- Cohn C, Joseph D (1962) Influence of body weight and body fat on appetite of “normal” lean and obese rats. Yale J Biol Med 34: 598-607.
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens NW, et al. (1996) Serum immunoreactive-leptin concentrations in normal-weight and obese humans. N Engl J Med 334:292-295.
- DeSouza CT, Araujo EP, Bordin S, Ashimine R, Zollner RL, et al. (2005) Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 146: 4192-4199.
- Myers MG Jr, Leibel RL, Seeley RJ, Schwartz MW (2010) Obesity and leptin resistance: distinguishing cause from effect. Trends Endocrinol Metab 21: 643-651.
- Gregor MF, Hotamisligil GS (2011) Inflammatory mechanisms in obesity. Annu Rev Immunol 29: 415-445.
- Thaler JP, Schwaetz MW (2010) Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology 151: 4109-4115.
- Myers MG, Cowley MA, Munzberg H (2008) Mechanisms of leptin action and leptin resistance. Annu Rev Immunol 70: 537-556.
- Shi X, Wang X, Li Q, Su M, Chew E, et al. (2013) Nuclear factor ?B(NF?B) suppresses food intake and energy expenditure in mice by directly activating the Pomc promoter. Diabetologia 56: 925-936.
- Howard JK, Flier JS (2006) Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab 17: 365-371.
- Munzberg H, Flier JS, Bjorback C (2004) Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology 145: 4880-4889.
- Howard JK, Cave BJ, Oskansen LJ, Tzameli I, Bjørbaek C, et al. (2004) Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med 10: 734-738.
- Mori H, Hanada R, Hanada T, Aki D, Mashima R, et al. (2004) Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med 10: 739-743.
- Reed AS, Unger EK, Olofsson LE, Piper ML, Myers MG Jr, et al. (2010) Functional role of suppressor of cytokine signaling 3 upregulation in hypothalamic leptin resistance and long-term energy homeostasis. Diabetes 59: 894-906.
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, et al. (2008) Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 135: 61-73.
- Ernst MB, Wunderlich CM, Hess S, Pachler M, Measoros A, et al. (2009) Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J Neurosci 29: 11582-11593.
- Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, et al. (2008) Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem 283: 14230-14241.
- White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, et al. (2009) HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab 296: 291-299.
- Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS,et al. (2006) Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med 12: 917-924.
- Picardi PK, Calegari VC, Prada PO, Moraes JC, Araújo E, et al. (2008) Reduction of Hypothalamic Protein Tyrosine Phosphatase Improves Insulin and Leptin Resistance in Diet-Induced Obese Rats. Endocrinology 149: 3870-3880.
- Thaler JP, Guyenet SJ, Dorfman MD, Wisse BE, Schwartz MW (2013) Hypothalamic inflammation: marker or mechanism of obesity pathogenesis? Diabetes 62: 2629-2634.
- Velloso LA, Schwartz MW (2011) Altered hypothalamic function in diet-induced obesity. Int J Obes (Lond) 35: 1455-1465.
- Yue JT, Lam TK (2012) Lipid sensing and insulin resistance in the brain. Cell Metab 15: 646-55.
- Youn JH (2014) Fat sensing and metabolic syndrome. Rev Endocr Metab Disord 15: 263-275.
- Milanski M, Degasperi G, Coope A, Morari J, Denis R, et al. (2009) Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 29: 359-370.
- Oh DY, Olefsky JM (2012) Omega 3 Fatty Acids and GPR120. Cell Metab 15: 564-565.
- Cintra DE, Ropelle ER, Moraes JC, Pauli JR, Morari J, et al. (2012) Unsaturated fatty acids revert diet induced hypothalamic inflammation in obesity. PLoS One 7: 30571.
- Obici S, Feng Z, Morgan K, Stein D, Karkanias G, et al. (2002) Central administration of oleic acid inhibits glucose production and food intake. Diabetes 51: 271-275.
- Milanski M, Arruda AP, Coope A, Ignacio-Souza LM, Nunez CE, et al. (2012) Inhibition of hypothalamic inflammation reverses diet-induced insulin resistance in the liver. Diabetes 61: 1455-1462.
- Moraes JC, Coope A, Morari J, Cintra DE, Roman EA, et al. (2009) High-fat diet induces apoptosis of hypothalamic neurons. PLoS One 4: 5045.
- Borg ML, Omran SF, Weir J, Meikle PJ, Watt MJ (2012) Consumption of a high-fat diet, but not regular endurance exercise training, regulates hypothalamic lipid accumulation in mice. J Physiol 590: 4377-89.
- Vinolo MA, Hirobara SM, Curi R (2012) G-protein-coupled receptors as fat sensors. Curr Opin Clin Nutr Metab Care 15: 112-116.
- Auvinen HE, Romijn JA, Biermasz NR, Pijl H, Havekes LM, et al. (2012) The effects of high fat diet on the basal activity of the hypothalamus-pituitary-adrenal axis in mice. J Endocrinol 214: 191-197.
- Serrano A, Pavon FJ, Tovar S, Casanueva F, Senaris R, et al. (2011) Oleoylethanolamide: Effects on hypothalamic transmitters and gut peptides regulating food intake. Neuropharmacology 60: 593-601.
- Postic C, Girard J (2008) Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest 118: 829-38.
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, et al. (2010) Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet 42: 937-948.
- Holland WL, Bikman BT, Wang LP , Yuguang G, Sargent KM, et al. (2011) Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid–induced ceramide biosynthesis in mice. J Clin Invest 121: 1858-1870.
- Benoit SC, Kemp CJ, Elias CF, Abplanalp W, Herman JP, et al. (2009) Palmitic acid mediates hypothalamic insulin resistance by altering PKC-θ subcellular localization in rodents. J Clin Invest 119: 2577-2589.
- Kleinridders A, Schenten D, Könner AC, Belgardt BF, Mauer J,et al. (2009) MyD88 signaling in the CNS is required for development of fatty acid-induced leptin resistance and diet-induced obesity. Cell Metab 10: 249-259.
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S , et al. (2009) Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab 9: 35-51.
- Kang YM, Ma Y, Zheng JP, Elks C, Sriramula S,et al. (2009) Brain nuclear factor-kappa B activation contributes to neurohumoral excitation in angiotensin II-induced hypertension. Cardiovasc Res 82: 503-512.
- Calegari VC, Torsoni AS, Vanzela EC, Araújo EP, Morari J, et al. (2011) Inflammation of the hypothalamus leads to defective pancreatic islet function. J Biol Chem 286: 12870-12880.
- Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, et al. (2011) Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci USA 108: 2939-2944.
- Wang YY, Lin SY, Chuang YH, Chen CJ, Tung KC, et al. (2011) Adipose proinflammatory cytokine expression through sympathetic system is associated with hyperglycemia and insulin resistance in a rat ischemic stroke model. Am J Physiol Endocrinol Metab 300: 155-163.
- Thaler JP, Choi SJ, Schwartz MW, Wisse BE (2010) Hypothalamic inflammation and energy homeostasis: resolving the paradox. Front Neuroendocrinol 31: 79-84.
- Ropelle ER, Flores MB, Cintra DE, Rocha GZ, Pauli JR, et al. (2010) IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKbeta and ER stress inhibition. PLoS Biol 8: 1000465.
- Hannun YA, Obeid LM (2008) Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 9: 139-150.
- Summers SA (2010) Sphingolipids and insulin resistance: the five Ws. Curr Opin Lipidol 21: 128-135.
- Holland WL, Brozinick JT, Wang LP, Hawkins ED, Sargent KM, et al. (2007) Inhibition of ceramide synthesis ameliorates glucocorticoid-, saturated-fat-, and obesity-induced insulin resistance. Cell Metab 5: 167-179.
- Kendel ER, Schwartz JH, Jessell TM (2000) Principles of neural science (4thedn). McGraw Hill, New York, USA.
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, et al. (2003) Obesity is associated with macrophage accumulation in adipose tissue. J Clin Inves 112: 1796-1808.
- Lumenberg CN, Saltiel AR (2011) Inflammatory links between obesity and metabolic disease. J Clin Invest 121: 2111-2117.
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, et al. (2012) Obesity is associated with hypothalamic injury in rodents and humans. J Clin Invest 122: 153-162.
- Yi CX, Al-Massadi O, Donelan E, Lehti M, Weber J, et al. (2012) Exercise protects against high-fat diet-induced hypothalamic inflammation. Physiol Behav 106: 485-490.
- Grayson BE, Levasseur PR, Williams SM, Smith MS, Marks DL, et al. (2010) Changes in melanocortin expression and inflammatory pathways in fetal offspring of nonhuman primates fed a high fat diet. Endocrinology 151: 1622-1632.
- Hidalgo J, Florit S, Giralt M, Ferrer B, Keller C, et al. (2010) Transgenic mice with astrocyte-targeted production of interleukin-6 are resistant to high-fat diet-induced increases in body weight and body fat. Brain Behav Immunol 24: 119-126.
- Tapia-González S, García-Segura LM, Tena-Sempere M, Frago LM, Castellano JM, et al. (2011) Activation of microglia in specific hypothalamic nuclei and the cerebellum of adult rats exposed to neonatal overnutrition. J Neuroendocrinol 23: 365-370.
- Yi CX, Tschop MH, Woods SC, Hofmann SM (2012) High-fat-diet exposure induces IgG accumulation in hypothalamic microglia. Dis Model Mech 5: 686-690.
- Lee D, Thaler JP, Berseth KE, Melhorn SJ, Schwartz MW, et al. (2013) Longer T2 relaxation time is a marker of hypothalamic gliosis in mice with diet-induced obesity. Am J Physiol Endocrinol Metab 304: 1245-1250.
- Guyenet SJ, Nguyen HT, Hwang BH, Schwartz MW, Baskin DG, et al. (2013) High-fat diet feeding causes rapid, non-apoptotic cleavage of caspase-3 in astrocytes. Brain Res 1512: 97-105.
- García-Cáceres C, Yi CX, Tschöp MH (2013) Hypothalamic astrocytes in obesity. Endocrinol Metab Clin North Am 42: 57-66.
- Morari J, Anhe GF, Nascimento LF, de Moura RF, Razolli D, et al. (2014) Fractalkine (CX3CL1) is involved in the early activation of hypothalamic inflammation in experimental obesity. Diabetes 63: 3770-3784.
- Valdearcos M, Xu AW, Koliwad SK (2015) Hypothalamic inflammation in the control of metabolic function. Annu Rev Physiol 77: 131-160.
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, et al. (2007) Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature 449: 228-232.
- Kumar RB, Aronne LJ (2014) Hypothalamic Inflammation: Is There Evidence for Human Obesity? Curr Obes Rep 3: 242-247.
- Briellmann RS, Kalnins RM, Berkovic SF, Jackson GD (2002) Hippocampal pathology in refractory temporal lobe epilepsy: T2-weighted signal change reflects dentate gliosis. Neurology 58: 265-271.
- Braffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, et al. (1988) Brain MR: pathologic correlation with gross and histopathology. 2. Hyperintense white-matter foci in the elderly. Am J of roentogenology 151: 559-566.
- Marshall VG, Bradley WG Jr, Marshall CE, Bhoopat T, Rhodes RH (1988) Deep white matter infarction: correlation of MR imaging and histopathologic findings. Radiology 167: 517-522.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) (2002) Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 106: 3143-3421.
- Buckman LB, Thompson MM, Moreno HN, Ellacott KL (2013) Regional astrogliosis in the mouse hypothalamus in response to obesity. J Comp Neurol 521: 1322-1333.
- Schur EA, Melhorn SJ, Oh SK, Lacy JM, Berkseth KE, et al. (2015) Radiologic evidence that hypothalamic gliosis is associated with obesity and insulin resistance in humans. Obesity (Silver Spring) 23: 2142-2148.
- Douglass JD, Dorfman MD, Thaler JP (2017) Glia: silent partners in energy homeostasis and obesity pathogenesis. Diabetologia 60: 226-236.
- Buckman LB, Hasty AH, Flaherty DK, Buckman CT, Thompson MM, et al. (2014) Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav Immun 35: 33-42.
- Berkseth KE, Guyenet SJ, Melhorn SJ, Lee D, Thaler JP, et al. (2014) Hypothalamic gliosis associated with high-fat diet feeding is reversible in mice: a combined immunohistochemical and magnetic resonance imaging study. Endocrinology 155: 2858-2867.
- André C, Guzman-Quevedo O, Rey C, Rémus-Borel J, Clark S, et al. (2016) Inhibiting Microglia Expansion Prevents Diet-Induced Hypothalamic and Peripheral Inflammation. Diabetes 66: 908-919.
- Buckman LB, Thompson MM, Lippert RN, Blackwell TS, Yull FE, et al. (2015) Evidence for a novel functional role of astrocytes in the acute homeostatic response to high-fat diet intake in mice. Mol Metab 4: 58-63.
- Sofroniew MV, Vinters HV (2010) Astrocytes: biology and pathology. Acta Neuropathol 119: 7-35.
- Yamanaka K, Chun SJ, Bollee S, Fujimori-Tonou N, Yamashita H, et al. (2008) Astrocytes as determinants of disease progression in inherited ALS. Nat Neurosci 11: 251-253.
- Osborn LM, Kamphuis W, Wadman WJ, Hol EM (2016) Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog Neurobiol 144: 121-141.
- Yang L, Qi Y, Yang Y (2015) Astrocytes control food intake by inhibiting AGRP neuron activity via adenosine A1 receptors. Cell Rep 11: 798-807.
- Chen N, Sugihara H, Kim J, Fu Z, Barak B, et al (2016) Direct modulation of GFAP-expressing glia in the arcuate nucleus bi-directionally regulates feeding. Elife 5: 18716.
- Kim JG, Suyama S, Koch M, Jin S, Argente-Arizon P, et al. (2014) Leptin signaling in astrocytes regulates hypothalamic neuronal circuits and feeding. Nat Neurosci 17: 908-910.
- Fuente-Martin E, García-Cáceres C, Argente-Arizón P, Diaz F, Granado M, et al. (2016) Ghrelin regulates glucose and glutamate transporters in hypothalamic astrocytes. Science Reports 6: 23673.
- Reiner DJ, Mietlicki-Baase EG, McGrath LE, Zimmer DJ, Bence KK, et al. (2016) Astrocytes Regulate GLP-1 Receptor-Mediated Effects on Energy Balance. J Neurosci 36: 3531-3540.
- García-Cáceres C, Quarta C, Varela L, Gao Y, Gruber T, et al. (2016) Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 166: 867-880.
- Douglass JD, Dorfman MD, Fasnacht R, Shaffer LD, Thaler JP (2017) Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol Metab 6: 366-373.
Citation: Kaur KK, Allahbadia G, Singh M (2018) Current Advances in Pathogenesis in Obesity: Impact of Hypothalamic Glioses. J Obes Weight Loss 3: 008.
Copyright: © 2018 Kulvinder Kochar Kaur, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Journal Highlights
© 2026, Copyrights Herald Scholarly Open Access. All Rights Reserved!