A Framework for Patient Stratification in Clinical Trials for Alzheimer’s Disease
*Corresponding Author(s):
Clara C CousinsDepartment Of Molecular And Cellular Biology, Harvard University, Cambridge, MA 02138, United States
Email:cousinsc15@gmail.com
Abstract
Drug discovery efforts in Alzheimer’s disease depend increasingly on systems to classify patients according to markers that reflect underlying pathological mechanisms. Such factors include prognostic indicators, such as genetic variants, as well as molecular markers and neuropathological subtypes with predictive or prognostic value. This review discusses how such markers might be used to identify probable responders to a drug and suggests a systematic framework within which clinical trials may classify patients according to disease state. Specifically, patients are likely to be clustered into homogeneous groups through a tiered system of stratification: first by the presence or absence of high-effect variants, then by functional similarity among less penetrant polymorphisms, levels of particular molecular biomarker classes, and neuroanatomical or symptomatic state.
Keywords
Alzheimer’s Disease; Molecular Markers
INTRODUCTION
Alzheimer’s disease (AD) culminates in a prominent phenotypic endpoint in the form of progressive cerebral neuronal atrophy. Nonetheless, recent clinical and pathophysiological evidence suggests that this common outcome may stem from a variety of causative pathways, each with variable contributions in particular subgroups [1]. Disease-modifying treatments are likely to rely on both early detection of disease and personalized interventions for particular AD variants, requiring markers with both predictive and prognostic value [2]. Accordingly, accounting for heterogeneity among AD patients has become a principal focus of drug discovery efforts in the field. This review discusses sources of heterogeneity in AD and presents a systematic framework for stratifying patient populations in clinical trials for drug candidates in preclinical and prodromal AD.
FACTORS CONTRIBUTING TO AD HETEROGENEITY
AD is a complex disease with competing etiological hypotheses. The strongest evidence for the dominant amyloid-b (Ab) theory stems from the identification of highly penetrant familial AD mutations in amyloid precursor protein (APP) and presenilin (PSEN) 1 and 2, which comprise the core components of a cleavage pathway leading to production of the Ab42 isomer [3]. Monomeric Ab42 can form fibrillary, extracellular aggregates in the form of amyloid plaques that characterize the later stages of the disease. However, it remains unclear whether the amyloid cascade is causative in all forms of AD, since improved cerebral Ab42 clearance does not always correlate with slowing of either neuronal atrophy or cognitive decline [4-6].
Vascular factors have also been implicated in AD pathogenesis. The high correlation of the apolipoprotein E4 (APOE4) variant with AD onset, with an odds ratio as high as 15:1, supports a vascular disease pathway, since mutant APOE has been shown to elicit an inflammatory response that disrupts endothelial structure within the microvasculature [7]. Similarly, polymorphisms in angiogenic regulators, such as VEGF and angiogenin, have been associated with sporadic AD [8]. Indeed, microvascular dysfunction appears in a significant proportion of AD cases, both cerebrally and systemically [9]. Perhaps most crucially, evidence from arteriopathic encephalopathies such as CADASIL and Glut1-induced epilepsy suggests that primary microvascular deficits can precede secondary neurodegenerative effects [10,11].
While these hypotheses do not account for all sources of pathological contribution, taken together they provide a useful framework for understanding disease heterogeneity through the so-called "multi-hit hypothesis” of AD [12,13]. In brief, vascular risk factors induce dysfunction in the microvasculature and disrupt systemic blood flow, leading to capillary hypoperfusion, accumulation of toxic metabolites, and, in combination with genetic factors, Ab deposition near the vasculature. All of these secondary outcomes combine to produce neuronal injury.
CLINICAL TRIAL DESIGN IN AD
An ideal patient stratification scheme should account for differing contributions from all relevant risk factors, which might include genetic predisposition, molecular indicators, and pathological stage. To date, AD clinical trials have fallen into one of four broad classes (Table 1). The earliest trials, beginning with tacrine (Pfizer) in 1987, were “general-participant,” enrolling patients with symptomatic diagnosis of mild-to-moderate AD [14]. The first immunotherapy trials, most notably AN-1792 (Elan) and solanezumab (Eli Lilly), fell into this class and uniformly failed due to inefficacy [15,16]. Assuming that these failures stemmed in part from late detection, subsequent studies shifted toward mild cognitive impairment (MCI) as a therapeutic target. While MCI trials led to two modestly symptom-improving drugs, an inability to distinguish pre-AD MCI reliably from other forms of dementia based on psychological evaluation has led to the discontinuation of MCI as a mainstream target indication for AD-modifying treatments [17].
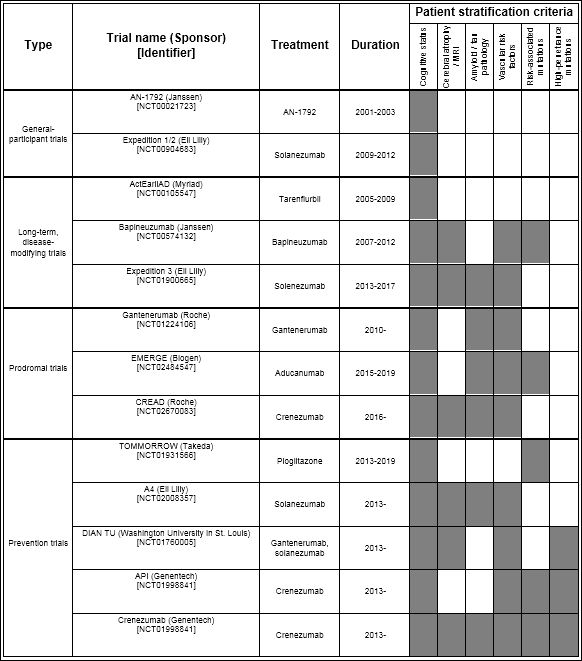
Table 1: Summary of notable clinical trials for Alzheimer’s disease from 2001 to 2019.
Notable phase 2/3 AD clinical trials are classified by study type and by stratification criteria. More recent trials tend to incorporate a greater variety of stratification tools.
The first trials that specifically addressed sources of AD heterogeneity began in 2001, enabled by the development of clinically relevant dementia ratings as long-term endpoints and effective methods of determining Ab load and APOE genotype [18]. This class of long-term, disease-modifying (LTDM) trials remains the de facto standard and includes several phase-3 anti-Ab trials, such as solanezumab and bapineuzumab [19,20]. However, the failure rate of this trial class has led many companies to consider two newer models that incorporate more sophisticated patient clustering methods. These trial structures impose stringent eligibility requirements, with smaller cohorts observed for longer durations. Prodromal trials, for instance, employ prognostic biomarkers like circulating Ab42 and amyloid positron-emission tomography (PET) scans as both entry criteria and indices of change [21]. In contrast, prevention studies such as the TOMMORROW trial and Dominantly Inherited Alzheimer Network Trial Unit (DIAN-TU) rely on predictive polymorphisms to identify and treat high-risk patients decades before the onset of symptoms [22,23].
A SYSTEMATIC FRAMEWORK FOR AD PATIENT STRATIFICATION
Prodromal and prevention trials represent the fastest growing classes of AD clinical studies, and their continued advancement is contingent on the development of efficient means of categorizing patients according to AD risk and severity. In combination, they suggest a structure for tiered patient stratification schemes in AD, which have already begun to emerge in recent prodromal trials (Figure 1) [24].
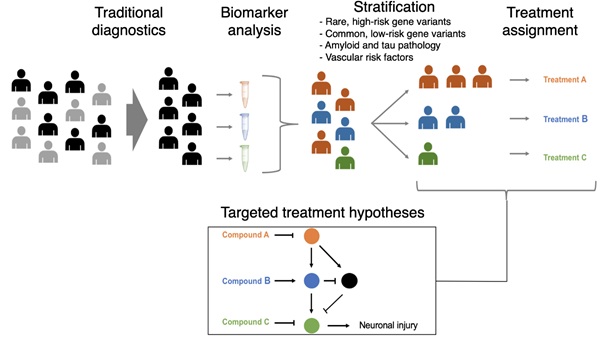
Figure 1: Biomarker analysis can supplement traditional diagnostics to stratify patients into more homogeneous treatment groups. The integration of clinical imaging, amyloid detection, vascular risk factors, and genotype can enable patient populations to be clustered by common disease mechanisms and ultimately targeted with pathway-modifying therapeutics.
The ultimate goal of a stratified approach to drug development in AD is to identify pharmacologically relevant disease subtypes at any stage where a clinically meaningful intervention can be made. As such, the approach will likely lend itself best to prevention trials for familial AD and preclinical and prodromal trials for sporadic AD, although LTDM trials may also benefit. After enrolment, primary patient clusters will first be generated according to a small number of highly penetrant genetic variants. These include mutations in APP and PSEN1/2, which implicate a dysfunctional APP cleavage pathway as a source of Ab deposition, as well as some APOE variants, which suggest a more significant contribution from vascular factors. These categories can be further refined according to risk-associated mutations in related pathways, which are vastly greater in number and collectively account for a larger proportion of AD heritability, although the genomic mechanisms of AD Neurodegeneration continue to be explored [25]. Accounting for the combination of rare variation with high effect size and common variation with low or even ambiguous effect size will enable genetically similar patient clusters to be defined before treatment administration, as has been successfully demonstrated in the development of immune checkpoint inhibitors for solid tumours. Pathways analysis can also facilitate the categorization of distinct risk-associated factors as “like” and “non-like,” allowing comparison between individuals with different haplotypes.
Accounting for genomic variation is crucial to the generation of scalable, homogeneous patient clusters for several reasons. First, genetic polymorphisms provide predictive value that cannot currently be obtained from molecular biomarkers. Furthermore, the lack of informative blood-based biomarkers means that determination of molecular biomarker levels is highly invasive, requiring either CSF extraction or PET-tracer injection [26]. Genetic analysis, while still restricted by cost, avoids the underlying problem of invasiveness and is therefore more likely to scale to large populations. Finally, it is thought that unidentified genetic factors explain a significant proportion of AD heritability [25]. These include both rare mutations with large effect size (such as polymorphisms in BIN1) and common but low-risk mutations, both of which are difficult to identify through traditional genome-wide association studies [27].
Once patients have been classified according to the presence or absence of dominant and risk-associated mutations, molecular biomarkers might provide some additional indication of disease state. Such markers are likely to include specific, cerebrospinal-fluid (CSF) based indicators like monomeric Ab42, phosphorylated tau-181, and total tau, as well as nonspecific, systemic markers such as micro vascular dysfunction [28]. PET imaging of cerebral amyloid deposition may also provide a valuable tool for further clustering, along with MRI analysis of regional atrophy in the brain. Finally, traditional cognitive profiles such as mini mental state evaluation (MMSE) may provide an additional dimension within each cluster [3]. Measures of non-cerebral pathology related to AD may also provide valuable information for resolving disease heterogeneity. For instance, video microscopy of the nail fold capillary (NFC) bed has demonstrated value as an inexpensive, non-invasive biomarker for AD dementia [29]. Such markers may provide insight into systemic contributions to vascular dysfunction in AD dementia.
It is important to note that using multiple tiered criteria for stratification does not necessarily limit the ultimate targetable population to restrictively small subsets of patients. Subtypes may share common targetable features (i.e., high cholesterol) downstream of distinct molecular causes. For instance, several studies have investigated the vascular component of AD by using anti-hypertensive drugs specifically in AD patients with hypertensive risk factors [30]. However, if a drug shows efficacy in a patient subgroup, pathways-analysis tools can be used to identify other groups that are likely to respond, as is common practice with new oncology therapies [31].
CONCLUSION
Development strategies for AD drug discovery are shifting toward the use of patient populations that are highly stratified with respect to underlying etiological factors. Current patient clustering methods rely on genetic classification, biomarker levels, and symptomatic differences, and future trials are likely to incorporate multiple features as predictive indicators of response. Specifically, a tiered system of patient classification may be most effective, using highly penetrant mutations, risk-associated variants, biomarker expression, and symptomatic classification. A system stratifying patients in these dimensions might also be supplemented by a scoring algorithm that weights different risk factors into a composite score, similar to the TOMMORROW trial’s biomarker risk assignment algorithm (BRAA). While retrospective application of this technique may be useful for identifying relevant biomarkers, clinical development of new drug candidates will ultimately require patient stratification in advance of treatment. Hopefully, as the toolkit for evaluating neurodegenerative heterogeneity expands, drug development in AD might begin to realize the potential of a precision-medicine approach.
REFERENCES
- Huang Y, Mucke L (2012) Alzheimer mechanisms and therapeutic strategies. Cell 148: 1204-1222.
- Gibbs RM, Lipnick S, Bateman JW, Chen L, Cousins HC, et al. (2018) Toward precision medicine for neurological and neuropsychiatric disorders. Cell Stem Cell 23: 21-24.
- Humpel C (2011) Identifying and validating biomarkers for Alzheimer’s disease. Trends Biotechnol 29: 26-32.
- Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, et al. (2013) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369: 341-350.
- Morris GP, Clark IA, Vissel B (2014) Inconsistencies and controversies surrounding the amyloid hypothesis of Alzheimer’s Disease. Acta Neuropathol Commun 2: 135.
- Bates KA, Verdile G, Li QX, Ames D, Hudson P, et al. (2009) Clearance mechanisms of Alzheimer’s amyloid-Β peptide: Implications for therapeutic design and diagnostic tests. Mol Psychiatry 14: 469-486.
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, et al. (2012) Apolipoprotein e controls cerebrovascular integrity via cyclophilin A. Nature 485: 512-516.
- Qin W, Jia X, Wang F, Zuo X, Wu L, et al. (2015) Elevated plasma angiogenesis factors in Alzheimer’s disease. J Alzheimer’s Dis 45: 245-252.
- Smith MM, Chen PCY, Li CS, Ramanujam S, Cheung ATW (2009) Whole blood viscosity and microvascular abnormalities in Alzheimer’s disease. Clin Hemorheol Microcirc 41: 229-239.
- Winkler EA, Nishida Y, Sagare AP, Rege S V, Bell RD, et al. (2015) GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18: 521-530.
- Salloway S, Hong J (1998) CADASIL syndrome: A genetic form of vascular dementia. J Geriatr Psychiatry Neurol 11: 71-77.
- Zhu X, Raina AK, Perry G, Smith MA (2004) Alzheimer’s disease: The two-hit hypothesis. Lancet Neurol 3: 219-226.
- Zhu X, Lee H gon, Perry G, Smith MA (2007) Alzheimer disease, the two-hit hypothesis: An update. Biochim Biochim Biophys Acta 1772: 494-502.
- Maltby N, Broe GA, Creasey H, Jorm AF, Christensen H, et al. (1994) Efficacy of tacrine and lecithin in mild to moderate Alzheimer’s disease: Double blind trial. BMJ 308: 879-883.
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, et al. (2005) Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64:1553-1562.
- Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, et al. (2014) Phase 3 Trials of Solanezumab for Mild-to-Moderate Alzheimer’s disease. N Engl J Med 370: 311-321.
- Allegri RF, Glaser FB, Taragano FE, Buschke H (2008) Mild cognitive impairment: Believe it or not? Int Rev Psychiatry 20: 357-363.
- McGhee DJM, Ritchie CW, Zajicek JP, Counsell CE (2016) A review of clinical trial designs used to detect a disease-modifying effect of drug therapy in Alzheimer’s disease and Parkinson’s disease. BMC Neurol 16: 92.
- Honig LS, Vellas B, Woodward M, Boada M, Bullock R, et al. (2018) Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med 378: 321-330.
- Salloway S, Sperling R, Fox NC, Blennow K, Klunk W, et al. (2014) Two phase 3 trials of Bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med 370: 322-333.
- Macklin EA, Blacker D, Hyman BT, Betensky RA (2013) Improved design of prodromal Alzheimer’s disease trials through cohort enrichment and surrogate endpoints. J Alzheimer’s Dis 36: 475-486.
- Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, et al. (2012) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367: 795-804.
- Burns DK, Chiang C, Welsh-Bohmer KA, Brannan SK, Culp M, et al. (2019) The TOMMORROW study: Design of an Alzheimer’s disease delay-of-onset clinical trial. Alzheimer’s Dement Transl Res Clin Interv 5: 661-670.
- Yassine HN (2017) Targeting prodromal Alzheimer’s disease: Too late for prevention? Lancet Neurol 16: 946-947.
- Van Cauwenberghe C, Van Broeckhoven C, Sleegers K (2016) The genetic landscape of Alzheimer disease: Clinical implications and perspectives. Genet Med 18: 421-430.
- Cacabelos R, Cacabelos P, Torrellas C, Tellado I, Carril JC (2014) Pharmacogenomics of Alzheimer’s disease: Novel therapeutic strategies for drug development. Methods Mol Biol 1175: 323-556.
- Lambert JC, Zelenika D, Hiltunen M, Chouraki V, Combarros O, et al. (2011) Evidence of the association of BIN1 and PICALM with the AD risk in contrasting European populations. Neurobiol Aging 32: 756.e11-756.e15.
- Olsson B, Lautner R, Andreasson U, Öhrfelt A, Portelius E, et al. (2016) CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol 15: 673-684.
- Cousins CC, Alosco ML, Cousins HC, Chua A, Steinberg EG, et al. (2018) Nailfold capillary morphology in Alzheimer’s disease dementia. J Alzheimer’s Dis 66: 602-611.
- Yasar S, Xia J, Yao W, Furberg CD, Xue QL, et al. (2013) Antihypertensive drugs decrease risk of Alzheimer disease: Ginkgo Evaluation of Memory Study. Neurology 81: 896-903.
- Schilsky RL (2010) Personalized medicine in oncology: The future is now. Nat Rev Drug Discov 9: 363-366.
Citation: Cousins HC, Cousins CC (2020) A Framework for Patient Stratification in Clinical Trials for Alzheimer’s Disease. J Alzheimers Neurodegener Dis 6: 033.
Copyright: © 2020 Henry C Cousins, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.