Alzheimer’s Disease Silent Killer of Memory: A Review on Pathological Mechanisms
*Corresponding Author(s):
Shamsher SinghDepartment Of Pharmacology, ISF College Of Pharmacy, Punjab, India
Tel:+91 9779980588,
Email:shamshersinghbajwa@gmail.com
Abstract
Alzheimer’s Disease (AD) is a Central Nervous System (CNS) related neurological disorder characterized by impairment of memory and finally turbulence in learning, reasoning, planning, language and perception. AD is a complex disease caused by an accretion of β - Amyloid (Aβ) plaques and neurofibrillary tangles formed by Tau amyloid fibrils linked with loss of synaptic plasticity and neurodegeneration. Apart from several other environmental factors, the rise in Oxidative Stress (OS), failure of mitochondria and Golgi plays a crucial role in neuronal dysfunctioning leads to neurodegeneration. However, oxygen is necessary for life and any imbalance between metabolism and excess Reactive Oxygen Species (ROS) generation leads to the origin of disorders such as Alzheimer’s disease. The ROS induced Mitochondrial (Mt) dysfunctions and excitotoxicity through over Ca2+ influx are two hallmarks behind neurodegeneration in AD. Still, there is no medicament presently available which can prevent further progression or decrease the symptoms of this disease. Some anti-oxidants were used but they were not effective due to their bioavailability problem into brain. The absence of effective therapeutic drugs and increased severity of AD day by day is a big challenge for against researchers. Therefore this review basically focused to explore the underlying pathological mechanism behind neuronal dysfunctioning and the various pathways involved in the pathogenesis of AD.
Keywords
ABBREVIATIONS
AD - Alzheimer’s Disease
AB - Amyloid Beta
NFTs - Neurofibrillary Tangles
CASP - Caspases
TNF-α - Tumor Necrosis Factor
APP - Amyloid Precursor Protein
ROS - Reactive Oxygen Species
IL - Interleukins
INTRODUCTION
Alzheimer's Disease (AD) is a slowly progressive neurological disorder characterized by impairment of memory and ultimately disturbances in learning, reasoning, planning, language, and perception [1]. It is also known as Senile Dementia of the Alzheimer Type (SDAT). Globally more than 45 million peoples are living with dementia, most suffering from AD. The prevalence of dementia will reach 75.6 million in 2030 and 135.5 million in 2050, of which 6.46% are from USA [2]. Many scientists believe that AD results from an increased oxidative stress due to the accumulation of a specific protein (beta-amyloid protein) in the brain that leads to neuronal cell death [3].
AD has been named after the German psychiatrist Alois Alzheimer who first described this disease in 1901 in Auguste D, a 50-year-old lady. Depending on its time of onset AD may be; Early-onset AD (usually affecting people aged 30 to 60 years) and Late-onset AD (this generally disturbs people over age 65 years) [4]. In AD patient may face problems in recognizing family and friends due to excessive neurodegeneration or neurotransmitter imbalance. The common symptoms in AD include alteration in mood, behavioral, hallucinations, and delirium. It usually starts slowly, silently and gets worse over time. With the advancement of disease physiological symptoms also alter like problems with language, alteration of mood, loss of inspiration and behavioral issues [5]. As a person's condition declines, she or he often withdraws from family and society, gradually body functions are lost leading to neuronal loss and death.
In addition, the accumulation of neurofibrillary tangles, free radical damage, low antioxidant enzymes level (Superoxide Dismutase (SOD) and catalase), mitochondrial failure have been observed in AD patients [6]. The increased oxidative damage to lipids and proteins with reduced activity of glutathione and other antioxidant enzyme are more localized to the neuronal junctions and correlate with the severity of the disease, suggesting to plays key role in AD-related neurodegenerative disorders [7]. The postmortem reports suggest that oxidative stress may be one of the prominent factors that is responsible for the initiation and development of AD. The exact pathophysiology of AD is still inadequately understood but mitochondria dysfunction and reduced level of pericyte are the progressive pathological factors [8].
Available evidence indicates that fragmentation of Golgi body increase Amyloid Precursor Protein (APP) trafficking and increase Amyloid Beta (Aβ) accumulation [9]. Joshi, et al., has suggested that the accumulation of Aβ peptides which leads to fragmentation of Golgi complex [10]. Nearly 70% risks of factors are of genetic background due to the involvement of various genes [11]. Additional risk factors include: person suffering from depression, accidental brain injury or hypertension. The major hallmark includes deposition of protein plaques (Tau proteins) and neurofibrillary tangles in the brain [12].
Nowadays there is no specific treatment available to prevent AD but the non-pharmacological measures like physical exercise as well as reducing body fat may decrease the risk of AD. Some of the antioxidants and vitamins from a long time are being under pre-clinical evaluation but these supplements cannot cure AD entirely or reverse its progression, though for the moment reduce symptoms [13]. Several drug therapies are available but they provide symptomatic relief to the patient suffering from this disease. The patients suffering from this disease become dependent on others for their daily life activities often increase a burden on the caregiver. Exercise programs are advantageous in the daily life and potentially improve outcomes. Antipsychotic agents are commonly used for the management of behavioral or psychotic problems due to dementia but not usually recommended due to their stumpy brain penetration and less therapeutic potential. Hence further studies are required to explore the hidden targets to achieve better drug therapy to secure life [14].
PATHOPHYSIOLOGICAL MECHANISMS IN ALZHEIMER’S DISEASE
Role of Amyloid Beta (Aβ) in neurodegeneration
In 2010 it was noted that the structural sequence on the oligomeric state of amyloid beta is still sparse. Low-temperature and low-salt conditions allowed isolating pentameric disc-shaped oligomers devoid of beta structure [24]. The amyloid beta after accumulation cause loss of neuronal functioning by inhibiting mitochondrial function, increase generation of ROS, further cause’s lipid peroxidation and the generation of a toxic aldehyde called 4-hydroxynonenal which in turn decreases ATPase’s activity, glucose transport and consequently neuronal loss occur. The β amyloid increases glutamate-dependent depolarization of the synaptic membrane which increases calcium influx dependent mitochondrial impairment [25]. Amyloid hypothesis stated that the fundamental cause of AD is extracellular Amyloid Beta (Aβ) deposits. This postulate has been evidenced by the position of the gene that regulates Amyloid Precursor Protein (APP) on chromosome 21, implying that individuals with Down's syndrome develop typical Alzheimer neuropathology because they produce too much Aβ lifelong [26]. Another fact that supports the above-mentioned postulate is that the people with trisomy 21 almost universally exhibit AD by 40 years of age [27]. Also, a specific form of Apolipoproteins (ApoE4) is the strongest known genetic risk factor for the late onset of AD [28]. Further, in the transgenic mice, it has been seen that those mice which express a mutant type of the human APP gene are more prone toward the development of fibrillary amyloid plaques and have more chances of AD. An experimental vaccine was used in early human trails to find out its effect for clearing the amyloid plaque but it was only effective for the clearance of plaque without significant effect on dementia. Researchers have been led to suspect non-plaque Aβ oligomers as the primary pathogenic form of Aβ. These noxious protein oligomers are also known to as Amyloid-Derived Diffusible Ligands (ADDLs). These oligomers easily bind to a cell membrane receptor of neurons and modify the structure of the synapse thereby distracting neuronal communication. A single receptor for Aβ protein oligomers may behave like prion protein, i.e. the same proteins which are associated with disease and the related human situation like Creutzfeldt-Jakob [29] (Figure 1).
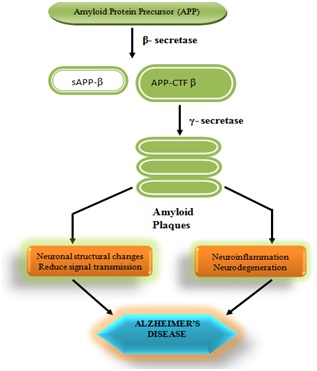
Golgi body and Amyloid beta protein: Available experimental reports suggest that the inhibitors of plasminogen activator inhibitor-1 decrease the plasma and brain Aβ levels in transgenic animals. Amyloid targeting immunotherapy can be done by bapineuzumab, gantenerumab etc. These monoclonal humanized antibodies mainly act on the CNS reduce Tau proteins level either by inhibiting their phosphorylation or clearance mechanism [30]. Exogenous antioxidants are being used like flavonoids, carotenoids and ubiquitous antioxidants show a powerful neuroprotective effect but have some bioavailability problems. The previous finding has reported that rutin, lutein, and curcumin also attenuates Aβ induced inflammation and reasonably inhibits β-secretase and AChE [31]. Normally the Aβ are matured and released by Golgi body but excessive accumulation or tauopathy cause fragmentation of this Golgi assembly that leads to loss of functioning [10].
Role of Tau protein in Alzheimer’s disease
The tau hypothesis, has proposed that any structural and functional tau protein abnormalities initiate’ disease cascade or disease like condition. Once the hyperphosphorylated tau accumulates inside nerve cell bodies, they cause disintegration of microtubules, destroy the cell's cytoskeleton that collapses the neuron transport system [34]. This may lead to malfunction of neurochemical communication among neurons that can cause neuronal loss or death [35]. Thus, according to Tau hypothesis it is believed that the abnormal aggregation of the tau protein is the cause of AD. Neurofibrillary tangles are thought to develop when an unknown trigger leads to the dissociation of tau from microtubules. Some studies revealed that hyperphosphorylated tau accumulates as tangles even when tau levels are decreased by transgenic suppression. This might be due to the action of tangles as a sink for tau intermediates that are predicted to have formed as part of the pathogenic process [33]. However, tangle formation is not associated directly with cognitive decline, which suggests that tau intermediates are the neurotoxic species; similar intermediates composed of other proteins such as Huntington or Aβ protein have been suggested to be neurotoxic elements in other diseases [36].
Tau-base targeted strategies prevent Alzheimer’s disease: Tau protein accumulation due to hyperphosphorylation loses microtubule binding and stabilizing role, contributing to neuronal deterioration [37]. Targeting tau phosphorylation is in basically needed to focus on Glycogen Synthase Kinase-3 (GSK3), one of the primary enzymes concerned in tau phosphorylation. A temperate improvement in mood (particularly apathy) and social interaction has been observed with cholinesterase inhibitors treatment [38]. Recent studies revealed that caffeine shows an inhibitory effect on Phosphodiesterease (PDE) enzyme and it also inhibits GSK3b. Microtubule stabilization criteria can be adopted in which the administration of paclitaxel (i.e. microtubule stabilizer) to tau-transgenic mice significantly improves fast axonal transport, microtubule density, and motor function. Epothilone D, is a potent inhibitor of active transporter which is also used as microtubule stabilizer is known for its good blood-brain barrier elimination [39]. Rojo, et al., has suggested that the lansoprazole and astemizole (both benzimidazole derivatives) bind with tau protein and in reducing the formation of neurofibrillary tangles. Enhancing tau degradation helps to augment the degradation of polymerized tau, thus declining its toxicity. Another, heat shock protein 90 (Hsp 90), also help to the denatured proteins, seems to play a role in preventing tau degradation [40]. Tau immunotherapy is successful for passive immunization with monoclonal antibodies decreases the phosphorylated tau molecules to reduce disease instances [41] (Figures 2&3).
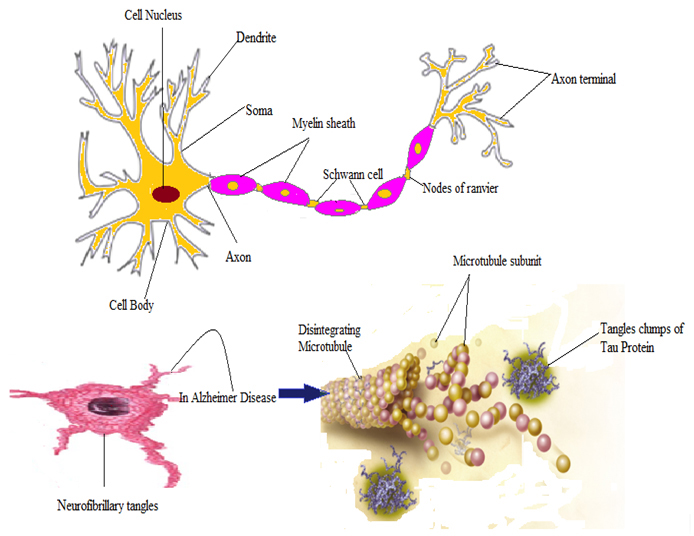 Figure 2: Formation of neurofibrillary tangles and tau protein clumps in Alzheimer’s disease.
Figure 2: Formation of neurofibrillary tangles and tau protein clumps in Alzheimer’s disease.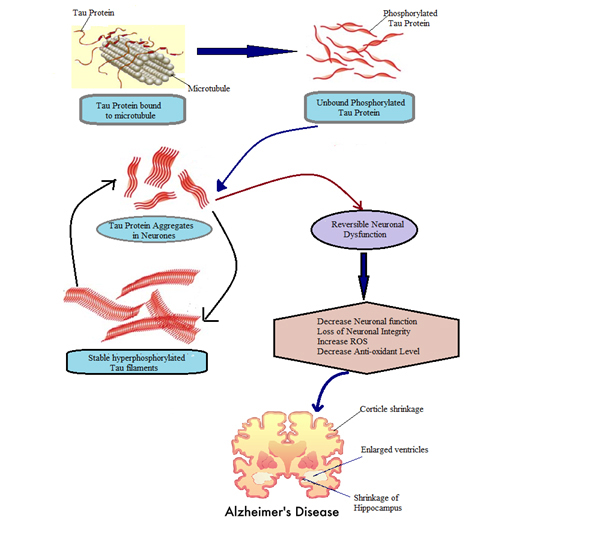 Figure 3: Role of Tau protein in Alzheimer’s disease.
Figure 3: Role of Tau protein in Alzheimer’s disease.Role of Pericyte in Alzheimer’s disease: Pericytes are individually situated in the neurovascular unit between endothelial cells of brain capillaries, astrocytes and neurons. Pericytes are cells in the Blood-Brain Barrier (BBB) which elevates the amyloid β-peptide (Aβ), cause tau pathology and neuronal loss that leads to progressive cognitive decline and dementia [42]. Most of the studies have shown that pericytes regulate key neurovascular functions including BBB formation and maintenance, regulation of capillary blood flow and clearance of toxic cellular by products necessary for normal functioning of the Central Nervous System (CNS). Abhay, et al., has observed that the pericytes control multiple steps of AD-like neurodegeneration pathogenic cascade in Aβ-precursor protein in mice (Figure 4).
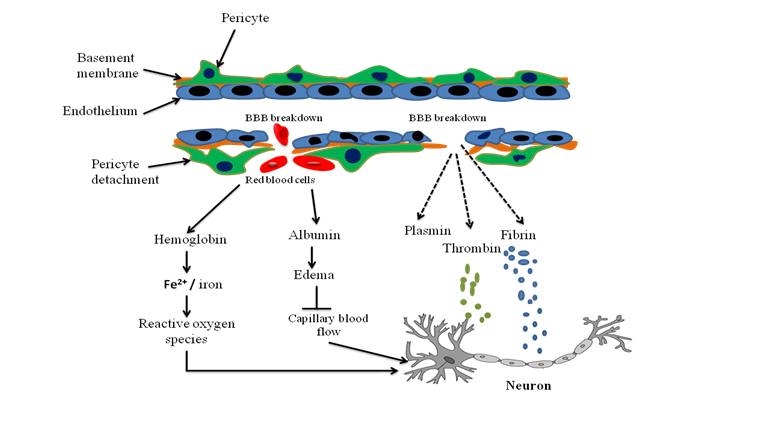 Figure 4: Role of pericyte in Alzheimer’s disease.
Figure 4: Role of pericyte in Alzheimer’s disease.Role of Oxidative Stress in Alzheimer’s disease
Increased oxidative stress and low cellular defense directly move the cells toward decay which can be reversed by enhancing antioxidants levels, such as glutathione. The detrimental effects of oxidative stress cause structural damage, cellular apoptosis which finally leads to necrosis. Production of ROS is a particularly destructive aspect of the cellular growth [47]. Some of the less reactive species (such as superoxide) can be converted into more destructive radical species by redox reactions with transition metals or other redox cycling compounds (including quinones) that can cause extensive cellular damage.
Similarly, the long-term effects are caused by damage to DNA which may be induced by ionizing radiation that is similar to oxidative stress and these lesions are mostly implicated in aging and cancer [48]. Some time the high-intensity radiation can cause single-base dents like 8-oxoguanine and thymine glycol and their biological effects have been studied extensively. Most of the oxygen-derived species responsible for oxidative stress cause necrosis are formed during normal cellular aerobic metabolism. The antioxidants are capable of destroying most of these oxygen derived species and any damage to cells is continually repaired. But under some harmful, conditions these species can cause necrosis leads to ATP depletion and finally delay the regeneration of the cells. Xanthine oxidase, NADPH oxidases, and cytochromes P450 are some of the other enzymes which are capable of producing superoxides [49]. Thus, to improve the cellular survival a balance must be necessary between production and consumption of free radical species. Disrupted mitochondrial respiratory ATP synthesis caused by oxidative stress leads to neurodegeneration followed by AD, Parkinson's disease and other neurodegenerative diseases [50].
Hypoxia exercise induces overproduction of (ROS) and RNS and alters redox balance. Infact, both acute and long-term hypoxic exposures have been shown to enhance the oxidative stress whereas increased oxidative stress has been observed in hypobaric as well as normobaric hypoxia (i.e., simulated altitude). Debevec, et al., has indicated that hypobaric hypoxia might induce higher oxidative stress levels than normobaric hypoxia [51] (Figure 5).
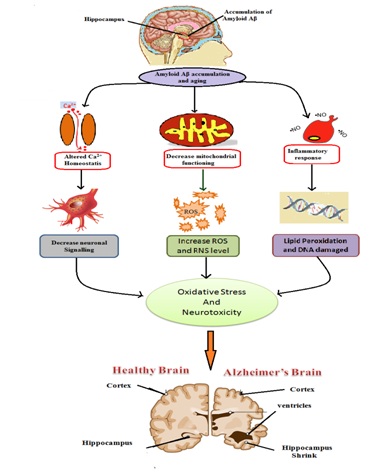 Figure 5:Effect of oxidative stress and mitochondrial failure in AD.
Figure 5:Effect of oxidative stress and mitochondrial failure in AD.Role of Caspase in Alzheimer’s Disease
 Figure 6: Process of activation of Executioner caspases.
Figure 6: Process of activation of Executioner caspases.It is well reported in various studies that the Amyloid Precursor Protein (APP) is a substrate for caspase-3-mediated cleavage, which may contribute to Aβ formation, synaptic loss, and the behavioral changes associated with AD. The principle hypothesis supporting this study was obtained using a site-directed Caspases-cleavage antibody to APP. In consideration to this report lot of studies have put forward and been able to demonstrate the caspases cleavage of fodrin in the AD brain as well as the activation of specific initiator and executioner caspases including caspase-3, 6, 8, and 9 [55] (Figure 7).
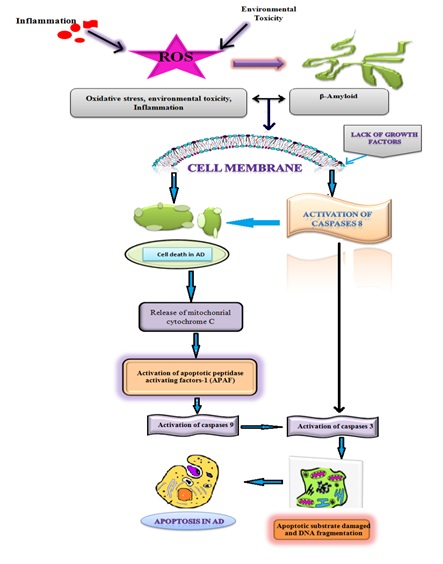 Figure 7: Role of Caspases in Alzheimer’s disease.
Figure 7: Role of Caspases in Alzheimer’s disease.Lastly, these studies provide lot of information which supports the activation of apoptotic pathways during the course of the disease progression in AD. Moreover, the role of caspases in AD is too much wider because the production of Aβ is also associated with their activation. Hence the number of studies suggest that caspases may be showing their proximal role in the disease mechanisms underlying AD including promoting Aβ formation as well as linking plaques to NFTs [56] (Figure 8).
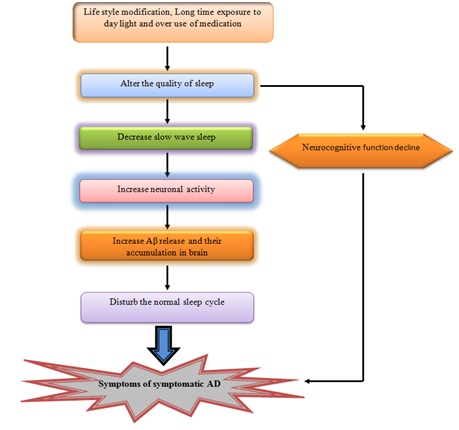 Figure 8: Sleep disturbances and Alzheimer disease.
Figure 8: Sleep disturbances and Alzheimer disease.Role of sleep changes in Alzheimer’s disease
Several reports have evidenced that sleep disruptions may be worse in patients with AD and due to this patient may lose the ability to stay asleep or keep alert during disease progresses [59]. Sleep problems may also increase the incidence of agitation among AD patient according to the results proofed by various reports [60]. Similarly, a term is used for this type of agitated behavior in AD that is "sundowning" mostly occurs in the evening schedule in various patients. This increased agitation behavior may be explained by sleepiness or may be more troublesome to caregivers during evening time [61]. But overall this creates a sleep apnea result in severe stress for caregivers and among AD patients which are on care therapy in nursing homes instead of at home. At initial stage this Sundown Syndrome is associated with increased stress, mental confusion and restlessness in patients due to which they are more prone to dementia that is commonly associated with AD [62].
The non-pharmacological factors which control the progression of AD are: firstly to make consistent sleeping, secondly change stressful lifestyle and lastly to do physical exercise. Physical activity is the best treatment for Alzheimer's, which also encourage patient for night sleep. Decrease the use of CNS stimulants like caffeine at night if a night's sleep is needed. Some evidence also supports the exogenous use of melatonin in combination with other antioxidants like lycopene or vitamin E can be used to induce sleep and preventing neurodegeneration [63]. Other pharmacological agents which are useful for the treatment of sleep deprivation induced or neurodegeneration induced AD are: Acetylcholinesterase inhibitor like donepezil which slows the cognitive decline in some patients but its major adverse effect is it can cause nighttime stimulation and is reported to promote nightmares in AD patients. Others like atypical antipsychotics and selective serotonin reuptake inhibitors are proved to be useful therapeutic agents [64].
CONCLUSION
AD is extremely complicated neurodegenerative disorder occurs due to the accumulation and overproductions of β-amyloid, tau formation by stressful lifestyle but the exact mechanism is still unknown. There are a number of molecular targets described which reflect the pathophysiology of AD but neurodegeneration have been speculated to occur through number of other factors also including environmental, genetic predisposition but oxidative stress and free radical generation are a prominent one. At present for the treatment of AD number of drugs like antioxidants and other pharmacological agents has been used but all provide symptomatic relief. So, there is a need for treatments which reduces the β-amyloid plaque formation or increases their elimination from CNS thereby reducing the oxidative stress. In this review, we have combined a number of molecular pathways involved in the pathogenesis of AD. Therefore either the inhibition of these pathways or the use of their antagonist may divert or decreases the progress of neurodegeneration in AD.
REFERENCES
- Woods LT, Ajit D, Camden JM, Erb L, Weisman GA (2016) Purinergic receptors as potential therapeutic targets in Alzheimer's disease. Neuropharmacology 104: 169-179.
- Guo T, Noble W, Hanger DP (2017) Roles of tau protein in health and disease. Acta Neuropathol 133: 665-704.
- Fang D, Zhang Z, Li H, Yu Q, Douglas JT, et al. (2016) Increased electron paramagnetic resonance signal correlates with mitochondrial dysfunction and oxidative stress in an Alzheimer’s disease mouse brain. J Alzheimers Dis 51: 571-580.
- Kudo T (2016) Basic Theory of Pharmacology for Alzheimer’s Disease. In Practical Pharmacology for Alzheimer’s Disease. Springer International Publishing 1-25.
- Godefroy O, Barbay M, Andriuta, D, Tir M, Roussel M (2017) Global Hypoactivity and Apathy. In Neuropsychiatric Symptoms of Cognitive Impairment and Dementia. Springer International Publishing 71-91.
- Schmitt K, Grimm A, Eckert A (2017) Amyloid-β-Induced Changes in Molecular Clock Properties and Cellular Bioenergetics. Front Neurosci 11: 124.
- Maluach AM, Misquitta KA, Prevot TD, Fee C, Sibille E, et al. (2017) Increased Neuronal DNA/RNA Oxidation in the Frontal Cortex of Mice Subjected to Unpredictable Chronic Mild Stress. Chronic Stress 1.
- Winkler EA, Sagare AP, Zlokovic BV (2014) The Pericyte: A Forgotten Cell Type with Important Implications for Alzheimer's Disease? Brain Pathol 24: 371-386.
- Huang S, Wang Y (2017) Golgi structure formation, function, and post-translational modifications in mammalian cells. F1000Res 6: 2050.
- Joshi G, Bekier MI, Wang Y (2015) Golgi fragmentation in Alzheimer’s disease. Front Neurosci 24: 340.
- Sathyan S, Barzilai N, Atzmon G, Milman S, Ayers E, et al. (2017) Association of anti-inflammatory cytokine IL10 polymorphisms with motoric cognitive risk syndrome in an Ashkenazi Jewish population. Neurobiol Aging 58: 238.
- Li K, Wei Q, Liu FF, Hu F, Xie AJ, et al. (2017) Synaptic Dysfunction in Alzheimer’s Disease: Aβ, Tau, and Epigenetic Alterations. Mol Neurobiol 55: 3021-3032.
- Watson R, Zibadi S (2017) Nutritional Modulators of Pain in the Aging Population (1st edn.). Academic Press, London, UK, Pg no: 314.
- Miarons M, Cabib C, Barón FJ, Rofes L (2017) Evidence and decision algorithm for the withdrawal of antipsychotic treatment in the elderly with dementia and neuropsychiatric symptoms. Eur J Clin Pharmacol 73: 1389-1398.
- Wildburger NC, Esparza TJ, LeDuc RD, Fellers RT, Thomas PM, et al. (2017) Diversity of amyloid-beta proteoforms in the Alzheimer’s disease brain. Scientific Reports 7.
- Willén K, Sroka A, Takahashi RH, Gouras GK (2017) Heterogeneous Association of Alzheimer’s Disease-Linked Amyloid-β and Amyloid-β Protein Precursor with Synapses. J Alzheimers Dis 60: 511-524.
- Wang X, Zhou X, Li G, Zhang Y, Wu Y, et al. (2017) Modifications and Trafficking of APP in the Pathogenesis of Alzheimer’s Disease. Front Mol Neurosci 10: 294.
- Stroo E, Koopman M, Nollen EA, Mata-Cabana A (2017) Cellular Regulation of Amyloid Formation in Aging and Disease. Front Neurosci 11: 64.
- Sweeney P, Park H, Baumann M, Dunlop J, Frydman J, et al. (2017) Protein misfolding in neurodegenerative diseases: implications and strategies. Transl Neurodegener 6: 6.
- Tarasoff-Conway JM, Carare RO, Osorio RS, Glodzik L, Butler T, et al. (2015) Clearance systems in the brain-implications for Alzheimer disease. Nat Rev Neurol 11: 457-470.
- Blonz ER (2017) Alzheimer’s Disease as the Product of a Progressive Energy Deficiency Syndrome in the Central Nervous System: The Neuroenergetic Hypothesis. J Alzheimers Dis 60: 1223-1229.
- Cardoso I, Santos LM, Alemi M (2016) Modulating Role of TTR in Aβ Toxicity, from Health to Disease Exploring New Findings on Amyloidosis 1-71.
- Economou NJ, Giammona MJ, Do TD, Zheng X, Teplow DB, et al. (2016) Amyloid β-protein assembly and Alzheimer’s disease: Dodecamers of Aβ42, but not of Aβ40 seed fibril formation. J Am Chem Soc 138: 1772-1775.
- Sengupta U, Nilson AN, Kayed R (2016) The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 6: 42-49.
- Kudryavtseva AV, Krasnov GS, Dmitriev AA, Alekseev BY, Kardymon OL, et al. (2016) Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 7: 44879-44905.
- Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8: 595-608.
- Karmiloff-Smith A, Al-Janabi T, D'Souza H, Groet J, Massand E, et al. (2016) The importance of understanding individual differences in Down syndrome. F1000Res 5.
- Foraker J, Millard SP, Leong L, Thomson Z, Chen S, et al. (2015) The APOE Gene is Differentially Methylated in Alzheimer's Disease. J Alzheimers Dis 48: 745-755.
- Cerasoli E, Ryadnov MG, Austen BM (2015) The elusive nature and diagnostics of misfolded Aβ oligomers. Front Chem 3: 17.
- Folch J, Petrov D, Ettcheto M, Abad S, Sánchez-López E, et al. (2016) Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural plasticity 1-15.
- Zhou X, Li Y, Shi X, Ma C (2016) An overview on therapeutics attenuating amyloid β level in Alzheimer's disease: targeting neurotransmission, inflammation, oxidative stress and enhanced cholesterol levels. Am J Transl Res 8: 246-269.
- Medina M, Hernández F, Avila J (2016) New Features about Tau Function and Dysfunction. Biomolecules 6: 21.
- Šimi? G, Babi? Leko M, Wray S, Harrington C, Delalle I, et al. (2016) Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6: 6.
- Brunden KR, Lee VM, Smith AB 3rd, Trojanowski JQ, Ballatore C (2017) Altered microtubule dynamics in neurodegenerative disease: Therapeutic potential of microtubule-stabilizing drugs. Neurobiol Dis 105: 328-335.
- Lepeta K, Lourenco MV, Schweitzer BC, Martino Adami PV, Banerjee P, et al. (2016) Synaptopathies: synaptic dysfunction in neurological disorders - A review from students to students. J Neurochem1 38: 785-805.
- Singh SK, Srivastav S, Yadav AK, Srikrishna S, Perry G (2015) Overview of Alzheimer’s Disease and Some Therapeutic Approaches Targeting Aβ by Using Several Synthetic and Herbal Compounds. Oxid Med Cell Longev.
- Alonso AD, Beharry C, Corbo CP, Cohen LS (2016) Molecular mechanism of prion-like tau-induced neurodegeneration. Alzheimers Dement 12: 1090-1097.
- Pandey MK, DeGrado TR (2016) Glycogen Synthase Kinase-3 (GSK-3)-Targeted Therapy and Imaging. Theranostics 6: 571-593.
- Ballatore C, Smith AB, Lee VMY, Trojanowski JQ, Brunden KR (2017) Microtubule-Stabilizing Agents for Alzheimer’s and Other Tauopathies. Alzheimer’s Disease 159-179.
- Watanabe H, Ono M, Saji H (2015) Novel PET/SPECT probes for imaging of tau in Alzheimer’s disease. Sci World J.
- Dai CL, Tung YC, Liu F, Gong CX, Iqbal K (2017) Tau passive immunization inhibits not only tau but also Aβ pathology. Alzheimers Res Ther 9: 1.
- Halliday MR, Rege SV, Ma Q, Zhao Z, Miller CA, et al. (2016) Accelerated pericyte degeneration and blood-brain barrier breakdown in apolipoprotein E4 carriers with Alzheimer’s disease. J Cereb Blood Flow Metab 36: 216-227.
- Sharma P, Jha AB, Dubey RS, Pessarakli M (2012) Reactive oxygen species, oxidative damage and antioxidative defense mechanism in plants under stressful conditions. Journal of Botany 1-26.
- Kaur R, Mehan S, Singh S (2018) Understanding multifactorial architecture of Parkinson’s disease: Pathophysiology to management. Neurol Sci.
- Sheng ZH (2014) Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol 204: 1087-1098.
- Perry G, Cash AD, Smith MA (2002) Alzheimer disease and oxidative stress. J Biomed Biotechnol 2: 120-123.
- Filomeni G, De Zio D, Cecconi F (2015) Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ 22: 377-388.
- Bariar B, Vestal CG, Richardson C (2013) Long-term effects of chromatin remodeling and DNA damage in stem cells induced by environmental and dietary agents. J Environ Pathol Toxicol Oncol 32: 307-327.
- Paravicini TM, Touyz RM (2008) NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care 31: 170-180.
- Yan MH, Wang X, Zhu X (2013) Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62: 90-101.
- Debevec T, Millet GP, Pialoux V (2017) Hypoxia-induced oxidative stress modulation with physical activity. Front Physiol 8: 84.
- Chang HY, Yang X (2000) Proteases for cell suicide: Functions and regulation of caspases. Microbiol Mol Biol Rev 64: 821-846.
- Gerl R, Vaux DL (2005) Apoptosis in the development and treatment of cancer. Carcinogenesis 26: 263-270.
- Pontillo A, Catamo E, Arosio B, Mari D, Crovella S (2012) NALP1/NLRP1 genetic variants are associated with Alzheimer disease. Alzheimer Dis Assoc Disord 26: 277-281.
- Kumasaka DK, Galvan V, Head E, Rohn TT (2009) Caspase cleavage of the amyloid precursor protein is prevented after overexpression of bcl-2 in a triple transgenic mouse model of Alzheimer’s disease. Int J Physiol Pathophysiol Pharmacol 1: 48-56.
- D’amelio M, Cavallucci V, Middei S, Marchetti C, Pacioni S, et al. (2011) Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer's disease. Nat Neurosci 14: 69-76.
- Jucker M, Walker LC (2011) Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Ann Neurol 70: 532-540.
- Bliwise DL (2004) Sleep disorders in Alzheimer's disease and other dementias. Clin Cornerstone 6: 16-28.
- Khachiyants N, Trinkle D, Son SJ, Kim KY (2011) Sundown syndrome in persons with dementia: an update. Psychiatry Investig 8: 275-287.
- Phillips B, Ancoli-Israel S (2001) Sleep disorders in the elderly. Sleep Med 2: 99-114.
- Volicer L, Harper DG, Manning BC, Goldstein R, Satlin A (2001) Sundowning and circadian rhythms in Alzheimer's disease. Am J Psychiatry 158: 704-711.
- Jelicic M, Bosma H, Ponds RW, Van Boxtel MP, Houx PJ, et al. (2002) Subjective sleep problems in later life as predictors of cognitive decline. Report from the Maastricht Ageing Study (MAAS). Int J Geriatr Psychiatry 17: 73-77.
- Cardinali DP, Furio AM, Brusco LI (2010) Clinical Aspects of Melatonin Intervention in Alzheimer’s Disease Progression. Curr Neuropharmacol 8: 218-227.
- Cacabelos R (2007) Donepezil in Alzheimer’s disease: From conventional trials to pharmacogenetics. Neuropsychiatr Dis Treat 3: 303-333.
Citation: Rana A, Awasthi A, Kumar D, Singh S, Singh S (2018) Alzheimer’s Disease Silent Killer of Memory: A Review on Pathological Mechanisms. J Alzheimers Neurodegener Dis 4: 017.
Copyright: © 2018 Arti Rana, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.