Emerging Role of Mitochondrial Function Enhancement in Mesenchymal Stem Cell Therapy
*Corresponding Author(s):
Soon Ha KimMitoImmune Therapeutics Inc., 13 F Glaston Bd. 108 Bongeunsa-ro, Gangman-gu, Seoul, Republic Of Korea
Tel:+82 269336726,
Fax:+82 269546728
Email:shakim@mitoimmune.com
Abstract
The impact of inflammatory environments on the impaired features of MSCs, such as loss of homeostasis and stem cell senescence, still remains unclear. In the paper titled “MIT-001 Restores Human Placenta-Derived Mesenchymal Stem Cells by Enhancing Mitochondrial Quiescence and Cytoskeletal Organization”, Yu et al., investigated the phenotypes of MSCs exposed to pro-inflammatory cytokines TNF-α and IFN-γ. They further tested whether the exposed MSCs are protected from senescence by MIT-001, a novel necrosis inhibitor and an anti-inflammatory molecule. The authors found that MSCs exposed to TNF-α and IFN-γ showed an abnormal cell shape, cytoskeletal organization, and senescence-associated secretory phenotypes. However, treatment with MIT-001 restored stem cell quiescence with improved mitochondrial distribution, reduced reactive oxygen species, and augmented mitochondrial energetics. The study suggests that enhancement of mitochondrial function restores MSCs quiescence and the application of MIT-001 mediated enhancement of mitochondrial function holds potential in future clinical therapeutics.
The Unmet Need in Mesenchymal Stem Cell (MSC) Therapy: Resolving Senescence
MSCs have the potential for self-renewal and multi-lineage differentiation and are hence being extensively explored for targeting inflammatory and immune-associated diseases through stem cell therapy. A meta-analysis evaluating the safety of MSC therapy and exploring the influence of heterogeneities of populations in clinical application over the past 15 years has confirmed the safety of MSC administration indifferent populations [1]. However, the promise of MSC-based treatments is currently limited because of MSCs undergoing senescence and consequently losing their potential. Resolving the issue of MSC senescence to allow them to continually proliferate and differentiate into multi-lineage mesenchymal cells is one of the greatest challenges in stem cell therapeutics and regenerative medicine and remains an unmet need.
Cytoskeletal Disorganization in Senescent MSCs
An increasing number of studies have shown a critical role of cytoskeleton in maintaining MSC shape, spreading, and stiffness, which in turn determine cell fate, differentiation, and function [2]. In aged MSCs, altered expression of actin cytoskeleton-associated proteins and a decreased actin turnover caused MSCs to be less responsive to biological and mechanical cues and reduced their migratory capacity. In Bone Marrow-derived Mesenchymal/Stromal Cells (BMSCs) obtained from a patient with Progressive Supranuclear Palsy (PSP), in which accumulation of hyper-phosphorylated Tau was prevalent, an alteration of microtubule association, a significant decrease in microtubule mass, and a hampered microtubule remodeling was observed during aging [3]. Remarkably, in Placenta-Derived Mesenchymal Stem Cells (PD-MSCs) showing higher morphological heterogeneity, and cytoskeletal disorganization, treatment with MIT-001 ameliorated unorganized microfilaments [4]. In this regard, the correction of actin cytoskeletal disorganization with MIT-001 seems to be a plausible approach to restore MSC functions.
Senescence-Associated Secretory Phenotype (SASP) in Senescent MSCs
The dynamic composition of SASP is known to lead to the transmission of MSC-senescence to surrounding cells, which eventually leads to the failure of stem cell therapy [5]. Interestingly, during in vitro expansion of umbilical cord blood MSCs, a cell population showed delayed cellular senescence by SASP activation and increased expression of p16, p21, and p53, implying a direct role of SASP in cell cycle arrest [6]. The inhibitory effect of MIT-001 on SASP was shown by the reduced expression of IL-1β and MCP-1 in PD-MSCs exposed to TNF-α and IFN-γ. In parallel, MIT-001 downregulated p16 expression, a cell cycle inhibitor, and improved senescent phenotypes [4]. These results suggest a role of MIT-001 in the inhibition of SASP in PD-MSCs, which will be beneficial for stem cell therapeutics.
Dysregulation Of Mitochondrial Function In Senecent MSCs
The relevance of mitochondrial function in MSCs has been overlooked because of the relatively low total mitochondrial mass in these cells band metabolic shift to glycolysis as the main energy source. However, a growing body of evidence has shown that mitochondrial Oxidative Phosphorylation (OXPHOS) and ATP generation is critical for the proliferation, differentiation, and survival of MSCs [7,8]. Thus, it is expected that elucidating the mechanisms maintaining mitochondrial homeostasis will pave the way for resolving the hurdles of MSC senescence and stem cell therapeutics. Many ongoing studies have focused on mitochondrial ROS, mitochondrial bioenergetics, mitochondrial dynamics, and Ca2+ buffering capacity, which are often altered in senescent MSCs. Understanding and targeting these will shed light on the potential of MSCs in therapy and avoid their senescence (Figure 1).
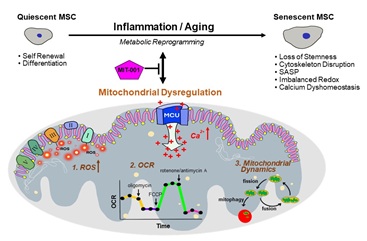 Figure 1: Altered mitochondrial homeostasis affects senescence in Mesenchymal Stem Cells (MSCs).
Figure 1: Altered mitochondrial homeostasis affects senescence in Mesenchymal Stem Cells (MSCs).
Mitochondrial ROS
Most of the energy gets generated through mitochondrial OXPHOS, with the involvement of the Electron Transport Chain (ETC) located at the inner mitochondrial membrane. The ETC is also regarded as the main source of intracellular ROS [9]. Although a certain level of ROS is essential for MSCs fate decision, evidence has demonstrated that higher levels of ROS in cells cause serious damage to biomolecules, including DNA mutations, lipid peroxidation, and protein modification [10]. In senescent MSCs, an increase in ROS is paralleled by a decrease in telomere maintenance and DNA repair after oxidative stress [11]. A critical role for ROS has been reported in the imbalanced MSC redox system [12]. Under the high ROS conditions, the activity of the transcription factor Nrf2, which regulates ROS scavenger enzymes, was reduced in senescent MSCs. In BMSCs from the patient with PSP, there was a significant increase in ROS and oxidative stress, coupled with a decrease in the cellular antioxidant glutathione and a high rate of mitochondrial degradation [3]. Therefore, reducing the levels of mitochondrial ROS seems to be of significance to enhance the activity of MSCs. Yu et al., confirmed this by showing an increase in ROS in PD-MSCs treated cytokines TNF-α and IFN-γ induced MSC senescence and showed that treatment with MIT-001 restored quiescence of MSCs [4].
Mitochondrial Oxygen Consumption Rate (OCR)
Mitochondrial OXPHOS and OCR can be used as indicators of mitochondrial function in physiology and pathophysiology. When cultured in normal versus high glucose media, MSCs showed higher OCR in the normal glucose medium, but more ROS accumulation in the cytosol and mitochondria with inflammatory cytokines IL-6 and TNF-α was found in the high glucose medium. Intriguingly, the upregulation of SOD2 significantly improved the OCR and reduced inflammatory cytokines in the high glucose medium [13]. These results suggest that the levels of ROS and antioxidant enzymes are the main regulators of OCR, although their contribution to OCR in MSCs still remains unknown. Human PD-MSCs exposed to TNF-α and IFN-γ showed increased spare respiratory capacity with a reduction of IL-β, IL-6, and MCP-1 [4]. Collectively, the assessment of mitochondrial energetics with the OCR may depict the functional activity of mitochondria and the potency of MSCs applicable for therapeutics. Moreover, enhancement of the spare respiratory capacity by MIT-001 could becritical for the improvement of the potential MSCs.
Mitochondrial Dynamics and Mitophagy
Cumulative data suggest that mitondrial dynamics contributeto the determination of MSC fate by varying the mitochondrialsize and shape [14,15]. For mitochondrial fission, dynamin-related protein Drp1is recruited from the cytosol to the mitochondria and mediates mitochondrial scission. Loss of Drp1 leads to inadequate mitochondrial fission, thus, inducing hyperfused mitochondria and mitochondrial DNA accumulation. For mitochondrial fusion, Mfn1 and Mfn2 mediate outer membrane fusion; followed byOPA1 mediated inner membrane fusion and remodeling of the cristae [16]. A lack of mitochondrial fusion leads to severe mitochondrial fragmentation and associated human diseases [17,18]. Interestingly, PD-MSCs exposed to TNF-α and IFN-γ showed increased expression of genes regulating fission and fusion processes, including Drp1, Mfn1, and Mfn2. Treatment with MIT-001 decreased the expression level of Drp1 and MFN1 mildly, whereas the decrease in Mfn2 expression was most significant. This indicates the potential of MIT -001 in correction of disrupted mitochondrial dynamics [4].
The loss of the autophagic ability of stem cells can cause the accumulation of unhealthy mitochondria; however, its effect on MSCs is yet to be elucidated [19]. Dysregulation of mitochondrial biogenesis in the BMSCs derived from the patient with PSP showed higher mitochondrial degradation and lower biogenesis [3], proving the critical role of mitophagy in cellular homeostasis.
Perspectives on Mitochondrial Calcium Regulation in MSCs
Increasing studies have weighed on mitochondrial Ca2+ buffering capacity and controlling of the ER shape and cytosolic Ca2+ signaling [20]. The release of ER-stored Ca2+ induced by pathological stimuli induces mitochondrial Ca2+ overload by uptake through the mitochondrial calcium uniporter. In a sepsis-induced heart dysfunction model, MSC-derived exosomes containing PINK-1 prevented mitochondrial Ca2+ overload and rescued cellular functions, underlying the importance of mitochondrial Ca2+ regulation in cellular homeostasis [21]. Suppression of mitochondrial Ca2+ overload by MIT-001 and its derivative in a myocardial reperfusion injury model has showed an increase in the survival of animals [22,23].
Mitochondrial Ca2+ overload generates ROS and contributes to oxidative stress in cells; reciprocally, the rise of ROS augments the Ca2+ surge. Hence, it is highly anticipated that mitochondrial Ca2+ overload may play a critical role in MSC senescence, of which ROS major regulator [24]. Therefore, regulation of mitochondrial Ca2+ uptake by MIT-001 and restoration of MSC quiescence holds promise for successful stem cell therapeutics.
Conclusion
Assessment of mitochondrial dysfunction for evaluating the potency of MSCs may be adopted for the therapeutic application of MSCs. Moreover, mitochondrial function enhancement by MIT-001 promises to be feasible for MSC therapy in clinical settings.
Acknowledgements
This research was supported by the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) and Korea Dementia Research Center (KDRC), funded by the Ministry of Health & Welfare and Ministry of Science and ICT, Republic of Korea (grant number: HU21C2273010021, SHK) and was supported in part by Basic Science Research Program through the National Research Foundation (NRF) of Korea funded by the Ministry of Education (grant number: 2018R1D1A1B07048900, JHY).
The authors appreciate Ms. Juri Na for her help in the illustration work.
Author’s Contribution
JHY conceptualized, designed, and wrote the manuscript. SHK conceptualized, designed, and reviewed the manuscript. Both authors read and approved the final manuscript.
Conflict of Interest
The authors work at MitoImmune Therapeutics Inc. A patent for MIT-001 is registered (USA patent no.: 9000028).
References
- Wang Y, Yi H, Song Y (2021) The safety of MSC therapy over the past 15 years: a meta-analysis. Stem Cell Res Ther 12: 545.
- Mathieu PS, Loboa EG (2012) Cytoskeletal and focal adhesion influences on mesenchymal stem cell shape, mechanical properties, and differentiation down osteogenic, adipogenic, and chondrogenic pathways. Tissue Eng Part B Rev 18: 436-444.
- Angelova PR, Barilani M, Lovejoy C, Dossena M, Vigano M, et al. (2018) Mitochondrial dysfunction in Parkinsonian mesenchymal stem cells impairs differentiation. Redox Biol 14: 474-484.
- Yu WD, Kim YJ, Cho MJ, Kim GJ, Kim SH, et al. (2021) MIT-001 Restores Human Placenta-Derived Mesenchymal Stem Cells by Enhancing Mitochondrial Quiescence and Cytoskeletal Organization. Int J Mol Sci 22.
- Zhou X, Hong Y, Zhang H, Li X (2020) Mesenchymal Stem Cell Senescence and Rejuvenation: Current Status and Challenges. Front Cell Dev Biol 8: 364.
- Kwon JH, Kim M, Um S, Lee HJ, Bae YK, et al. (2021) Senescence-Associated Secretory Phenotype Suppression Mediated by Small-Sized Mesenchymal Stem Cells Delays Cellular Senescence through TLR2 and TLR5 Signaling. Cells 10: 63.
- Pattappa G, Thorpe SD, Jegard NC, Heywood HK, Bruijn JD, et al. (2013) Continuous and uninterrupted oxygen tension influences the colony formation and oxidative metabolism of human mesenchymal stem cells. Tissue Eng Part C Methods 19: 68-79.
- Zhao L, Hu C, Zhang P, Jiang H, Chen J (2019) Mesenchymal stem cell therapy targeting mitochondrial dysfunction in acute kidney injury. J Transl Med 17: 142.
- Kukreja RC, Kontos HA, Hess ML, Ellis EF (1986) PGH synthase and lipoxygenase generate superoxide in the presence of NADH or NADPH. Circ Res 59: 612-619.
- Atashi F, Modarressi A, Pepper MS (2015) The role of reactive oxygen species in mesenchymal stem cell adipogenic and osteogenic differentiation: a review. Stem Cells Dev 24: 1150-1163.
- Yi J, Luo J (2010) SIRT1 and p53, effect on cancer, senescence and beyond. Biochim Biophys Acta 1804: 1684-1689.
- Stavely R, Nurgali, K (2020) The emerging antioxidant paradigm of mesenchymal stem cell therapy. Stem Cells Transl Med 9: 985-1006.
- Sen S, Domingues CC, Rouphael C, Chou C, Kim C et al. (2015) Genetic modification of human mesenchymal stem cells helps to reduce adiposity and improve glucose tolerance in an obese diabetic mouse model. Stem Cell Res Ther 6: 242.
- Ren L, Chen X, Chen X, Li J, Cheng B, et al. (2020) Mitochondrial Dynamics: Fission and Fusion in Fate Determination of Mesenchymal Stem Cells. Front Cell Dev Biol 8: 580070.
- Ziegler DV, Wiley CD, Velarde MC (2015) Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 14: 1-7.
- Liu YJ, McIntyre RL, Janssens GE, Houtkooper RH (2020) Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech Ageing Dev 186: 111212.
- Zhou Y, Carmona S, Muhammad A, Bell S, Landeros J, et al. (2019) Restoring mitofusin balance prevents axonal degeneration in a Charcot-Marie-Tooth type 2A model. J Clin Invest 129: 1756-1771.
- Alavi MV, Fuhrmann N (2013) Dominant optic atrophy, OPA1, and mitochondrial quality control: understanding mitochondrial network dynamics. Mol Neurodegener 8: 32.
- Lin Q, Chen J, Gu L, Dan X, Zhang C, et al. (2021) New insights into mitophagy and stem cells. Stem Cell Res Ther 12: 452.
- Yoast RE, Emrich SM, Zhang X, Xin P, Arige V, et al. (2021) The Mitochondrial Ca(2+) uniporter is a central regulator of interorganellar Ca(2+) transfer and NFAT activation. J Biol Chem 297: 101174.
- Zhou Q, Xie M, Zhu J, Yi Q, Tan B, et al. (2021) PINK1 contained in huMSC-derived exosomes prevents cardiomyocyte mitochondrial calcium overload in sepsis via recovery of mitochondrial Ca(2+) efflux. Stem Cell Res Ther 12: 269.
- Thu VT, Kim HK, Long le T, Lee SR, Hanh TM, et al. (2012) NecroX-5 prevents hypoxia/reoxygenation injury by inhibiting the mitochondrial calcium uniporter. Cardiovasc Res 94: 342-350.
- Hwang IC, Kim JY, Kim JH, Lee JE, Seo JY, et al. (2012) Therapeutic Potential of a Novel Necrosis Inhibitor, 7-Amino-Indole, in Myocardial Ischemia-Reperfusion Injury. Hypertension 71: 1143-1155.
- Peng, TI, Jou MJ (2010) Oxidative stress caused by mitochondrial calcium overload. Ann N Y Acad Sci 1201: 183-188.
Citation: Yang JH, Kim SH (2022) Emerging Role of Mitochondrial Function Enhancement in Mesenchymal Stem Cell Therapy. J Stem Cell Res Dev Ther 7: 089.
Copyright: © 2022 Jeong-Hee Yang, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.