Enhancement of sAPP as a Therapeutic Strategy for Alzheimer’s and Other Neurodegenerative Diseases
*Corresponding Author(s):
Varghese JohnDepartment Of Neurology, Drug Discovery Laboratory, Easton Center For Alzheimer’s Disease Research, Geffen School Of Medicine, University Of California, Los Angeles, California, United States
Tel:+1 3102064345,
Email:vjohn@mednet.ucla.edu
Abstract
Soluble, secreted Amyloid Precursor Protein-α (sAPPα), a product of α-secretase (ADAM10) cleavage of Full Length-APP (FL-APP), is a trophic factor critical for synaptic complexity and maintenance. As cleavage at the α-site of APP precludes the β-site cleavage that is the first step in Amyloid β (Aβ) production, enhancing sAPPα production may not only support and restore neuronal health, but may also decrease the generation of anti-trophic Aβ. Over-production or reduced clearance of Aβ is a hallmark of Alzheimer’s Disease (AD), and recent findings suggest it also plays a role in other neurodegenerative diseases and neurological conditions, such as Amyotrophic Lateral Sclerosis (ALS), Cerebral Amyloid Angiopathy (CAA), and Traumatic Brain Injury (TBI). Yet decades of focus on Aβ-lowering strategies alone including passive and active immunotherapy and γ-secretase and BACE1 (BACE) inhibition have yet to yield positive clinical results. Clinical trials of several BACE inhibitors are underway in AD patients, and although there is optimism about this strategy, there are also concerns about mechanism-based side-effects of these drugs. A truly effective therapy would not only slow the degenerative process underlying onset and progression of the disease, it should also restore healthy neuronal function. It is very likely this will comprise combination therapy utilizing more than one drug or intervention. Molecules that enhance sAPPα may be a safe, effective component of a multi-modal therapeutic approach to AD and other neurodegenerative diseases, and have the potential to increase neuronal health by providing trophic support and disrupting neurodegenerative mechanisms.
Keywords
INTRODUCTION
Therapeutics for Alzheimer’s Disease (AD)
More recent approaches to AD therapeutic development include re-purposing the anti-epileptic drug levetiracetam to address the seizure-like activity manifest in many AD patients [7,8], use of anti-diabetic drugs including intranasal insulin [9,10], and development of BACE inhibitors [11-13], including our own APP-selective BACE inhibitors [14], to name a few. It is hoped that some of these new approaches will provide benefit in AD, but it is very likely that truly effective treatment for AD will require multiple therapeutics working in concert - similar to the approach used for AIDS therapy - to address the many deficits in the disease. One component of this multi-modal therapy should restore and promote normal neuronal function, rather than just arrest a single deleterious process underlying the disease. It is our hypothesis and that of others that enhancement of trophic peptide sAPPα, or the activity of the enzyme ADAM10, could play this key role in therapy.
Aβ plaques and the amyloid hypothesis in AD
Some of the strongest support for the amyloid hypothesis of AD comes from identification of mutations that lead to familial forms of Alzheimer’s Disease (FAD). These mutations are found in APP, presenilin (executor of γ cleavage) 1 or 2 genes, and the ADAM10 gene [23]. All of these mutations result either in increased Aβ production or increased Aβ1-42 production relative to other Aβ species, which in turn leads to Early Onset Alzheimer’s Disease (EOAD). Furthermore, an A673T mutation in APP at the β-cleavage site has recently been described that protects against AD and cognitive decline in elderly persons in the absence of AD [24]. Subsequent studies showed this mutation decreased β cleavage of APP, and was associated with a slight reduction in aggregation of Aβ1-42 peptides [25].
The great majority of AD cases, however, are sporadic and do not result from inherited mutations. These are classified as Late Onset Alzheimer’s Disease (LOAD). Genetics can play a role in LOAD, as possession of either one or two Apolipoprotein E ε4 alleles (ApoEε4) confers an increased risk for the development of AD [26]. Apolipoproteins bind Aβ and affect transport, aggregation and clearance as well as influence synaptic plasticity, cell signaling, lipid transport and metabolism, and neuroinflammation [27]. Because there is close association with Aβ accumulation in ApoEε4 individuals and cognitive decline frequently leading to development of AD, this is further support for the amyloid hypothesis.
The manifestation of AD-like cognitive changes in the absence of amyloid plaques - if neurofibrillary tangles are present - is considered to be a tauopathy rather than AD. Some subjects with cognitive changes in the absence of AD-like pathology, as determined by amyloid PET imaging but with degenerative changes in 18fluorodeoxyglucose PET scans and hippocampal volume, have also been identified. In one study, these Suspected Non-Alzheimer Pathology (SNAP) individuals comprised approximately one third of patients diagnosed with cognitive decline with age and most were found to have cerebrovascular disease or synucleinopathy [28]. Other conditions such as Lewy body dementia, depression, or multiple sclerosis may lead to manifestation of AD-like cognitive changes, but in the absence of amyloid pathology, are not defined as AD.
APP processing
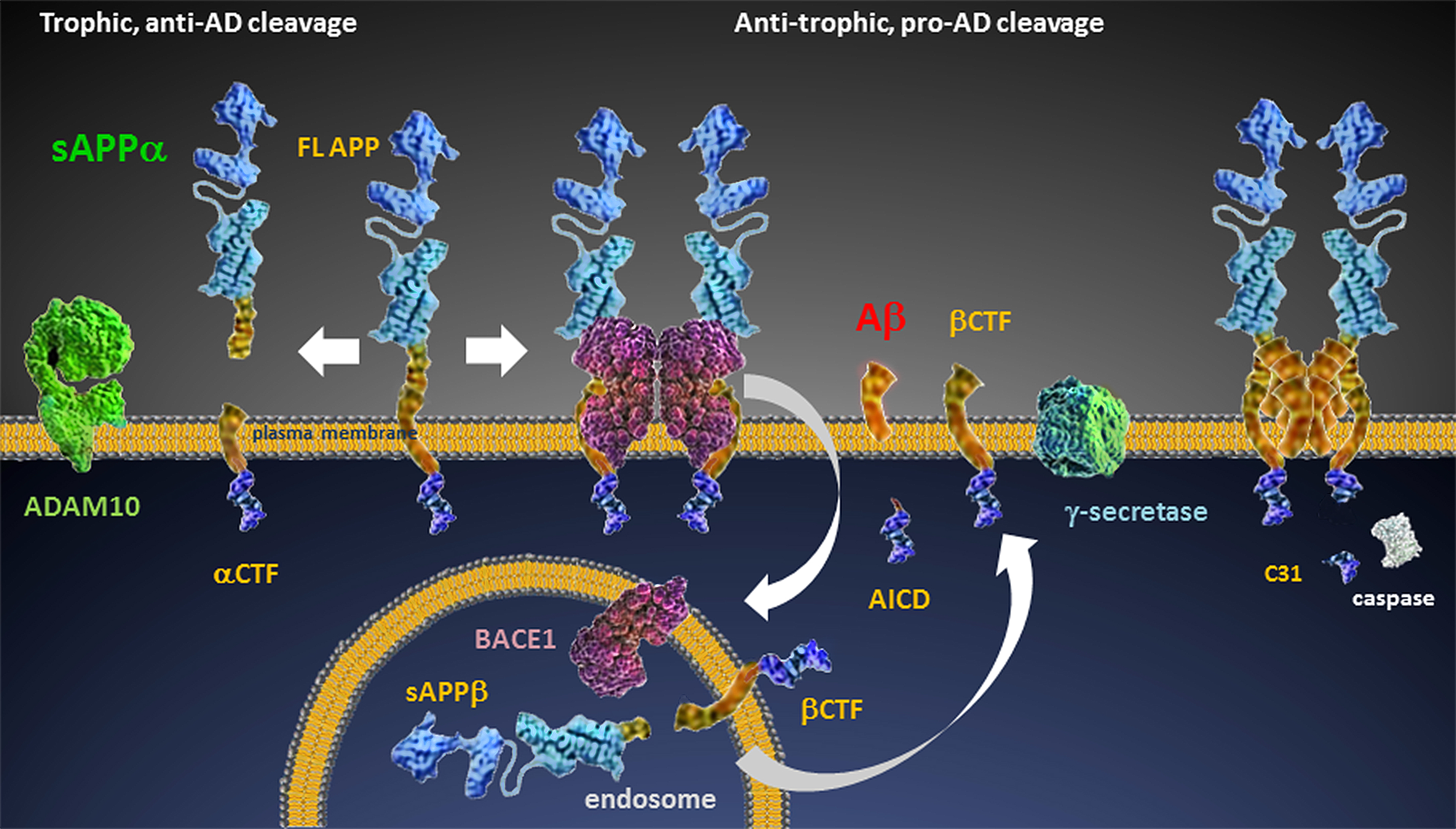
The pro-cognitive role of sAPPα is clear. In primary neuronal culture, sAPPα decreases excitability [37] and in synaptosomes, it increases synaptic elements [38]. The correlation of sAPPα to synaptic plasticity and maintenance is well-established in vivo. In Anderson et al., [39] a positive correlation was shown between performance in spatial memory tasks and CSF sAPPα in young and aged rats. Intracerebroventricular (ICV) treatment with sAPPα improved both motor and cognitive function in mice subjected to Traumatic Brain Injury (TBI), another pathological condition resulting in increased β-pathway processing of APP [40]. Recently, it was shown that acute sAPPα administration can rescue LTP in conditional APP/APLP knockout mice [41]. In humans, CSF sAPPα levels were seen to correlate positively with better cognitive performance [42] as determined by IQ, verbal ability, visuospatial function, immediate memory, episodic memory and various aspects of attention.
Mutations in ADAM10 and at APPα- and β-cleavage sites alter AD risk
Kaden et al., [44] were the first to identify and characterize the K16N mutation, a lysine-to-asparagine substitution localized to the α-secretase cleavage site which causes early onset autosomal dominant dementia. The mutation increased Aβ toxicity and dramatically diminished α-cleavage and therefore α-CTF and sAPPα generation, resulting in levels 40-50% lower than those seen with APP wild type.
Conversely, in the search for low-frequency variants in the APP gene with significant effects on AD risk, Jonsson et al., [24] found the A673T coding mutation that protects against AD as well as age-related cognitive decline in the absence of AD. This substitution is adjacent to the β-site, and results in a reduction in Aβ production and a reduction of approximately 32% in the sAPPβ/sAPPα ratio in vitro [25]. While the effects of these mutations appeared largely to be manifest in a reduction in sAPPβ and βCTF production, they do suggest reduction of the sAPPβ/sAPPα ratio by enhancement of sAPPα may have a similar effect. Interestingly, a recessive mutation at the same site - A673V - seen in an Italian family [45] causes enhanced Aβ production and fibril formation only when homozygous. When heterozygous, co-expression of wildtype APP and wildtype Aβ destabilizes aggregates and decreases toxicity, making this mutation either advantageous or disadvantageous with respect to AD, depending upon zygosity. This finding also suggests a new therapeutic strategy for destabilizing toxic aggregates in AD [46].
ApoEε4, SirT1 and sAPPα in AD
Sirtuins are NAD-dependent deacetylases that affect longevity and have a myriad of metabolic and stress-tolerance functions. Significant decreases in SirT1 levels in parietal cortex in AD patient tissue have been reported, and these decreases were closely correlated with duration of symptoms and tau accumulation [50]. We found similar SirT1 decreases in temporoparietal cortex from AD patients; specifically, SirT1 - but not SirT2 or SirT6 - was significantly decreased by more than 60%. In addition, Kumar et al., [51] revealed a pronounced decline in SirT1 serum concentration in AD and MCI, as well as a more moderate decline in age-matched cognitively normal individuals.
Our studies reveal that ApoEε4 triggers a reduction in sAPPα levels by inhibiting the proteolysis of APP at the α-site and by reducing transcription of SirT1. As SirT1 has previously been shown to activate transcription of ADAM10 [52] and thus increase levels of neuroprotective sAPPα [53,54], one can conclude that a decrease in SirT1 expression as a result of the presence of ApoEε4 is likely to lead to decreased sAPPα levels. In further support of a role for ApoEε4 effects on SirT1 expression and ultimately sAPPα production, we showed that increasing SirT1 expression in the presence of ApoEε4 restores sAPPα levels in vitro by co-transfecting A172 cells with both ApoEε4 and SirT1 and identifying increases in sAPPα of 10% (1:1) or 20% (1:2) as compared to ApoEε4 transfection alone [47].
As a result of these findings, we added SirT1 enhancement as a target for our in vitro screens to identify new small molecules for potential development as AD therapeutics. This resulted in our identification of brain-permeable small molecules that increase SirT1, sAPPα, and cell survival in vitro (manuscript in preparation). In our ongoing studies, we plan to test these molecules in vivo, as well as screen a larger compound library to identify additional novel SirT1 enhancers.
TrkA overexpression decreases sAPPα
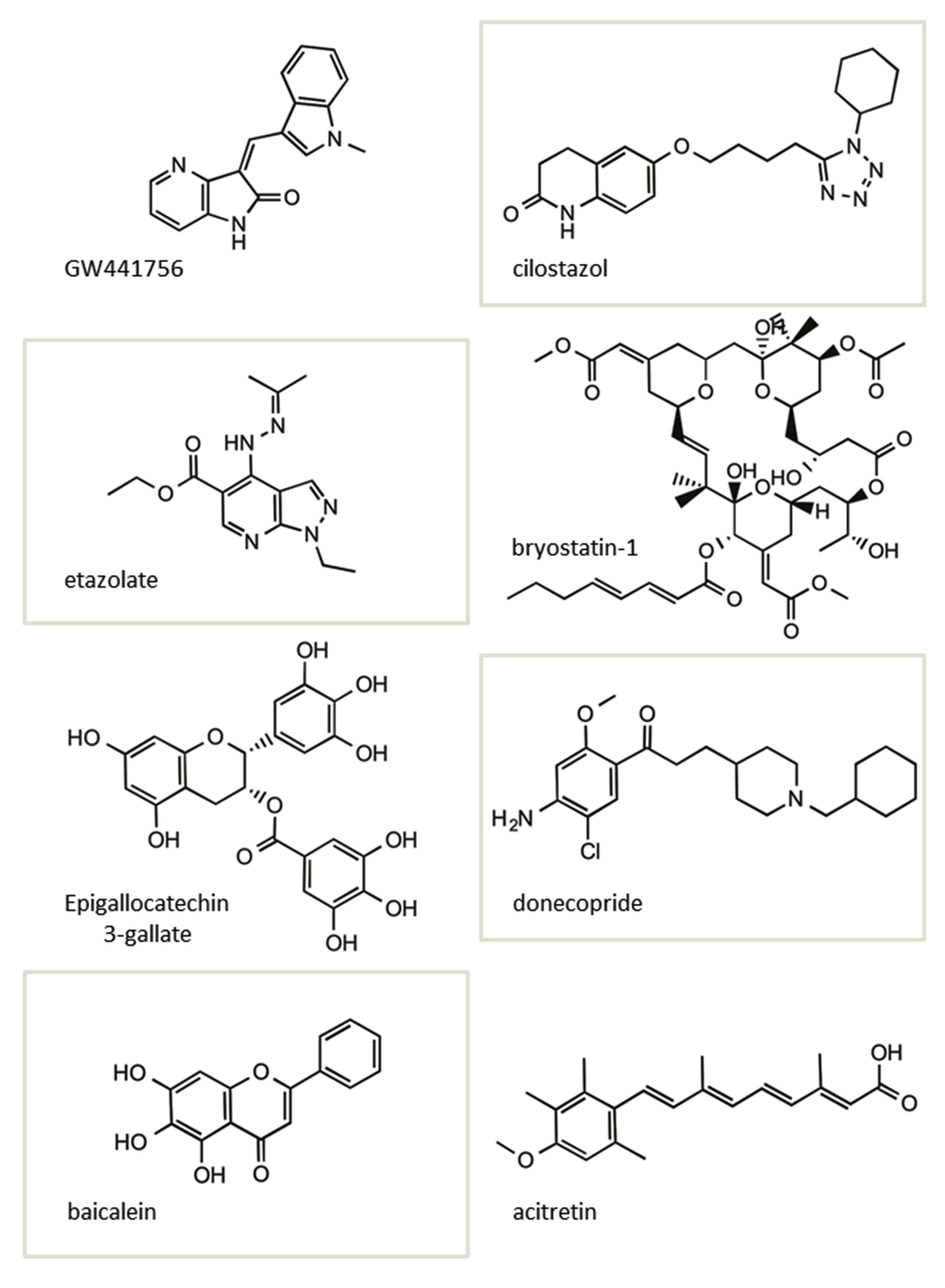
As ADDN-1351 was not brain penetrant and could not be tested in vivo, we tested the known TrkA inhibitor GW441756 in the J20 PDAPP mouse model of AD [56], and saw that it increased sAPPα, suggesting TrkA inhibition - rather than NGF activation - as a novel therapeutic approach to AD.
The finding that an inhibitor of the Nerve Growth Factor (NGF) receptor TrkA exerts anti-AD effects is, at first, counter-intuitive as it has been shown that reduction of TrkA-NGF interaction is associated with AD. In AD, however, Basal Forebrain Cholinergic Neurons (BFCN) degenerate due, in part, to impaired retrograde transport of NGF-TrkA complexes from BFCN targets. This may lead to accumulation of these complexes, over-activation, and C31 production. Hence, these complexes may have a paradoxical effect: under normal physiological conditions NGF signaling through TrkA may result in inhibition of the amyloidogenic pathway, but in AD, impaired retrograde transport of NGF-TrkA complexes may be deleterious.
sAPPα is a potent inhibitor of BACE
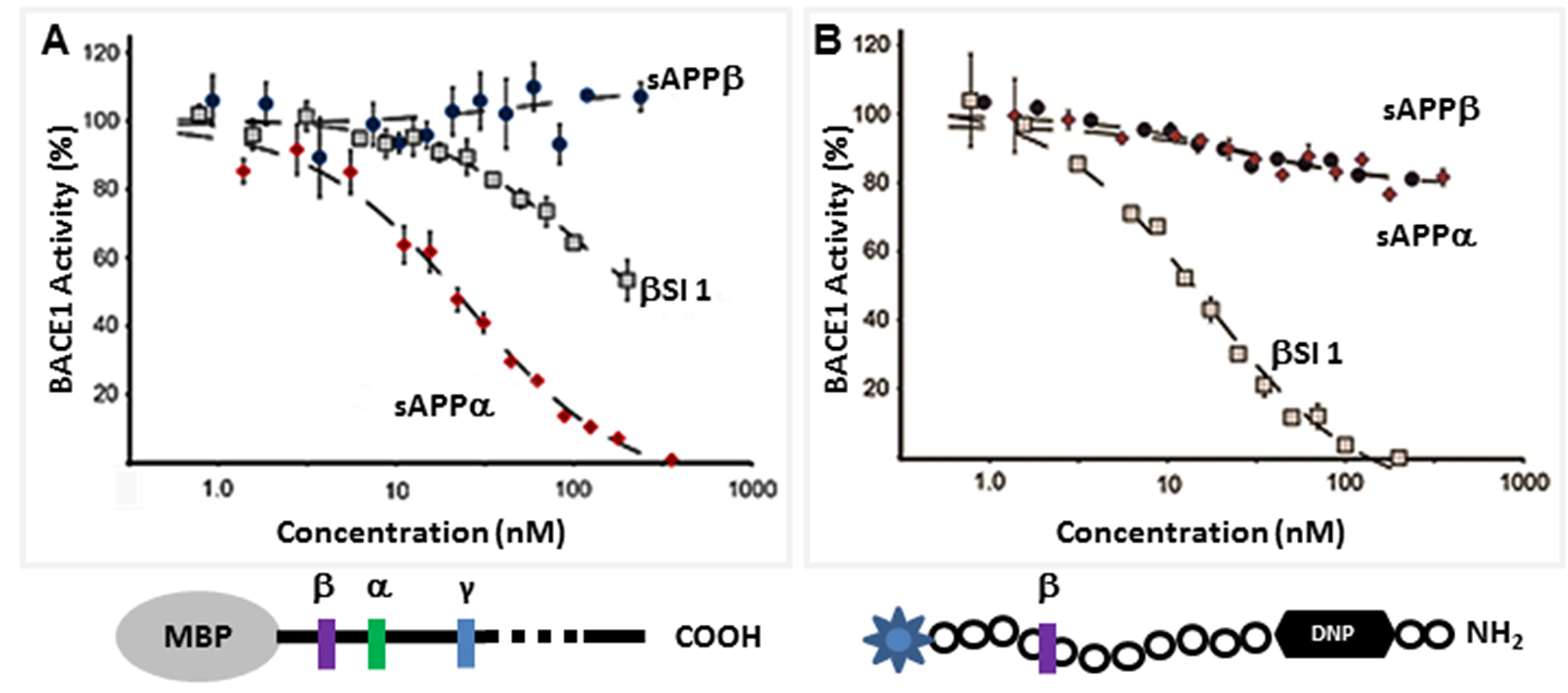
The direct inhibition of BACE by sAPPα indicates that it acts as an endogenous inhibitor ligand that can selectively regulate the proteolysis of APP. Our biochemical analysis reveals that sAPPα is an allosteric BACE inhibitor, showing an inhibitory profile similar to that of an exosite binding antibody [57]. Additionally, our Small-Angle X-ray Scattering (SAXS) analysis shows that sAPPα adopts a conformation distinct from the slightly shorter non-inhibitor sAPPβ [32], and this conformational difference may explain their differing effects. BACE inhibition is an appealing strategy for AD therapeutic development. There are advanced BACE inhibitors currently in Phase 3 clinical trials, such as the Merck BACE inhibitor MK8931 [58]. All are directly active site-binding BACE inhibitors that interact with the catalytic dyad of the enzyme. It is hoped that these compounds will perform well in the clinic, but it is not improbable that they may be associated with unwanted side effects due to off-target cleavage inhibition of non-APP substrates. The APP-selective allosteric inhibition of BACE cleavage of APP may be associated with fewer risks for side effects.
sAPPα or ADAM10 enhancers in AD
Meta analysis of more than 20 in vivo studies in the AD model mice showed that F03 induces highly significant increases in sAPPα (Figure 3C), decreases Aβ1-42, and increases the sAPPα/Aβ1-42 ratio. F03 repeatedly increased cognitive performance in mice in the pre-plaque stage and, strikingly, it was able to improve cognition as determined using the Novel Object Recognition (NOR) testing paradigm in old J20 AD model mice with extensive pre-existing Aβ plaque pathology (Figure 3D). This improvement in working object memory, as well as improvement in spatial memory as determined by Novel Location Recognition (not shown), was closely associated with increases in sAPPα (Figure 3E) as no significant decreases in Aβ were seen (Figure 3F). The effects of F03 in vivo were seen at low, human-equivalent doses used to treat Post-Operative Nausea and Vomiting (PONV), and as F03 is known to have a good safety profile, the drug is now in a clinical Phase 1b/2a in AD patients in Australia.
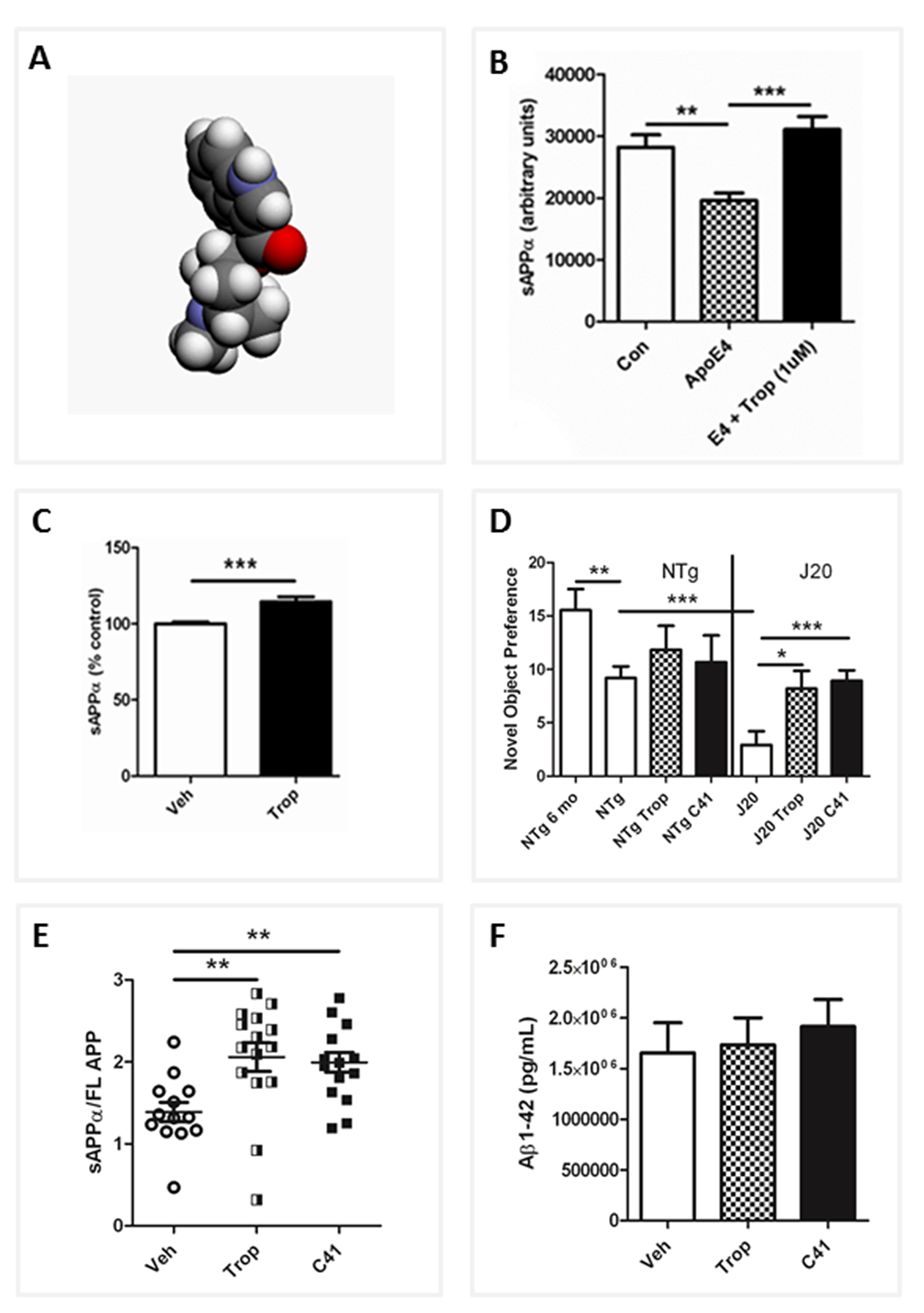
In our ongoing studies, we have generated and tested more than thirty F03 analogs and have noted the sAPPα-enhancing effects come largely from the 5-HT3 antagonism, while the Aβ-lowering effects appear to arise from α7nAChR agonism. Surface Plasmon Resonance (SPR) shows F03 also interacts directly with APP and this, combined with multifunctional receptor interactions, seems to be critical for efficacy. In addition, F03 has been reported as an anti-inflammatory agent [60-62]. As chronic inflammation may be a factor contributing to the onset of AD [63], this drug may be of even greater utility in the treatment or prevention of AD.
Others are also identifying sAPPα enhancers. Lee et al., found that cilostazol attenuates Aβ production by increasing ADAM10 activity via SirT1-coupled Retinoic Acid Receptor-β (RARβ) activation in N2a cells expressing human APP Swedish (Swe) mutation [64]. Interestingly, in this study, SirT1 overexpression in N2a Swe cells also elevated ADAM10 and sAPPα levels.
Generally, retinoic acids are known to up regulate ADAM10 expression and/or activity [52]. Acitretin, a synthetic retinoid that is an approved drug for psoriasis, increases the ADAM10 gene expression. In a pilot Phase 2 clinical study in AD patients, acitretin was shown to significantly increase the sAPPα levels in CSF after a short period of treatment [65]. A longer term study on a larger patient cohort with this drug is planned.
Other molecules known to increase ADAM10 expression and/or activity include muscarinic agonists, neuropeptides such as Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP), Protein Kinase C (PKC) activators, phosphatidylinositol 3-kinase, cAMP and calcium [66,67].
Ginsenoside Rh2, a ginseng derivative, was found to improve learning and memory in a mouse model of AD [68], and in vitro increased soluble sAPPα. Qiu et al., also used live-cell labeling to show plasma membrane APP levels increased and APP endocytosis decreased, and that this effect was likely due to a reduction in lipid raft levels. A green tea-derived polyphenolic compound (-)-epigallocatechin-3 gallate reduced Aβ in vitro and in vivo in an AD mouse model, in part by activation of estrogen receptor-α/phosphatidylinositide 3-kinase/protein kinase B signaling and by increasing ADAM10 processing [69].
Donecopride [70] a dual (h) 5-HT4R partial agonist also promotes sAPPα release and exerts pro-cognitive effects at 0.3 and 1 mg/kg in a mouse model of AD.
Baicalein, a flavonoid that modulates γ-Aminobutyric Acid (GABA) type A receptors, also increases sAPPα [71]. in vitro, baicalein significantly reduced the production of β-Amyloid (Aβ) by increasing APP α-processing. AD mice treated with baicalein for eight weeks showed enhanced APP α-secretase processing, reduced Aβ production, and reduced AD-like pathology together with improved cognitive performance.
The Protein Kinase C (PKC) activator bryostatin-1, a macrolide lactone extract from a bryozoan species, has been shown to be effective in increasing sAPPα levels while reducing Aβ40 and 42 in AD mouse models [72], and is currently in clinical trials for AD.
The structures of several of the small molecules shown to increase sAPPα in vitro and in vivo described above are shown in (Figure 4). Interestingly, two of these molecules, bryostatin-1 and acitretin, in addition to F03 (Figure 3A), have advanced into clinical testing in MCI and AD patients.
sAPPα and Cerebral Amyloid Angiopathy (CAA)
Enhancement of sAPPα in Amyotrophic Lateral Sclerosis (ALS)
In Yoon et al., [76] it was revealed that Aβ interacts with Super Oxide Dismutase 1 (SOD1) resulting in a reduction in SOD activity, an increase in oxidative stress, and the compromised mitochondrial function that is characteristic of ALS [77,78]. In ALS model mice, APP and Aβ are both upregulated in spinal cord [74] and overexpression of Aβ has been shown to accelerate the onset of motor impairment [79]. Furthermore, the studies of Herman et al., [80] revealed that increasing Aβ42 increased the Tar-DNA binding Protein (TDP43) inclusions found in the majority of ALS patients. Pathologically high Aβ concentrations exacerbate glutamate excitotoxicity at the Neuro-Muscular Junction (NMJ), a major contributor to motor neuron loss in ALS [81]. Reduction of Aβ production and trophic support by sAPPα enhancement may, therefore, be a new effective therapeutic strategy for ALS, at least as part of multimodal therapy. It has been shown that ICV treatment with a monoclonal antibody that blocks the β-secretase cleavage site on APP results in reduction of sAPPβ and Aβ levels, and delays disease onset and deterioration in the pre-symptomatic stage of the disease in ALS mice [82].
sAPPα has several neuroprotective and/or trophic effects that may specifically be of benefit in ALS. Through receptor binding, it alters cyclic-GMP (cGMP) production and activates a cGMP-dependent Protein Kinase (PKG), promoting activation of the nuclear transcription factor NF-kB. sAPPα has been found to protect neurons from proteasomal stress by inhibiting the stress-triggered pro-apoptotic c-Jun N-terminal Kinase (JNK)-signaling pathway. This may be of great utility in those cases of ALS wherein proteasomal stress is increased by accumulation of TDP43 and Fused-in-Sarcoma (FUS). This also suggests sAPPα enhancement may be of utility, at least as part of multi-model therapy, in Frontotemporal Dementia (FTD) where in TDP43 deposits are also found.
Some of the strongest evidence that sAPPα is implicated in ALS was revealed in Steinacker et al., wherein low CSF sAPPα levels were found to be tightly correlated with rapid disease progression. SirT1 expression was recently shown to ameliorate disease progression in a mouse model of ALS [83-85], indicating that sAPPα and/or SirT1enhancement might both be effective as part of ALS therapy.
sAPPα, Traumatic Brain Injury (TBI) and stroke
The upregulation of APP may lead to concomitantly increased sAPPα and Aβ, indicating that both may play a role in response to TBI [40]. And while there may be a protective role for increased Aβ production acutely post-TBI [90-92], ongoing increases in Aβ production are likely deleterious and increase the risk for later development of AD. The restoration of trophic pre-injury APP processing may improve outcome. Based on this hypothesis, Thornton et al., [93] introduced sAPPα ICV post-trauma to rats with induced TBI. The results included significantly improved motor outcome in rotorod testing, reduction of the number of apoptotic neuronal perikarya in hippocampus and cortex, and reduced axonal injury within the corpus callosum, revealing it to be a promising therapeutic strategy for TBI.
The benefits of increasing sAPPα after a hypoxic event such as TBI or stroke may partially be attributed to its inhibition of BACE. As we posit in Peters-Libeu et al., [32] the inhibition of BACE by sAPPα may be part of an evolutionarily-conserved hypoxia response pathway. Hypoxia-Inducible Factor (HIF)-1 has been shown to upregulate both BACE and APP expression in zebra fish [94] and in mammalian cell culture [95]. Similarly, production of both BACE and APP have been shown to be up-regulated in response to hypoxia in the developing rat brain and in mature rats [96], leading to increased production of Aβ peptide. In addition, suppression of HIF-1 has been shown to decrease BACE production and increase sAPPα production [97].
An exaggerated inflammatory response also contributes to poorer outcome post-TBI, in part due to a reduction in sAPPα production. Therefore sAPPα may act as a modulator of inflammation. Siopi et al., [98] studied the effects of the α-secretase activator etazolate on acute and post-TBI outcome in a mouse model. Within a therapeutic window of two hours, a single dose of etazolate reduced inflammation and edema, and improved memory and locomotion, with these effects closely associated with restoration of sAPPα levels.
sAPPα, sleep and melatonin
CONCLUSION
The dominant paradigm in AD research posits accumulation of Aβ in brain as the key biochemical event underlying the development of AD, and thus is a primary target for drug development. Yet targeting Aβ production and/or clearance has, to date, resulted in clinical failure in treatment of AD. This could very well be due to inadequate target engagement by the therapeutics or drug-related side effects rendering the treatment modality ineffective. Successful clearance of amyloid by antibody treatment such as with bapineuzumab has been shown to be associated with Amyloid-Related Imaging Abnormalities-Edema/Effusion (ARIA-E), apparently reflecting microhemorrhage. Similarly, off-target effects of the γ-secretase inhibitors due to inhibition of cleavage of non-APP substrates such as notch 1 resulted to significant side effects for this approach. Other failures may be due to trial participant selection or screening, particularly when participants with AD-symptomology do not have amyloid pathology. PET amyloid imaging is being used to eliminate this issue.
Timing of treatment may also likely be a critical factor leading to clinical trial failure. By the time an AD diagnosis is made, there is already significant neuronal death and neurofibrillary tangle formation, which Aβ-lowering alone cannot reverse. Therefore, to test the amyloid hypothesis, it would be ideal to commence anti-Aβ or amyloid treatment before significant damage has occurred, but in a patient population where the onset of amyloid pathology is almost certain. In a current study that is addressing these issues, individual members of families in Colombia expressing a genetic mutation resulting in increased Aβ production are being treated pre-symptomatically with the humanized antibody crenezumab. In addition, the ongoing anti-amyloid treatment in the asymptomatic AD “A4” study - a 3-year prevention trial in PET-positive 65-85 year-old participants with the Aβ antibody solanezumab-targets older individuals with normal cognition but at risk of developing sporadic AD. In this case, appropriate study participants are identified by pre-study amyloid PET imaging.
BACE inhibitors are currently in clinical trials and still hold great promise, but again may be limited in use by off-target side effects. Even if these inhibitors provide some benefit, as we hope, it is likely combination therapy will be necessary to achieve a truly significant effect. As a result, there is an urgent need to identify new approaches for the treatment of AD, and it is possible that targeting sAPPα enhancement will be advantageous, improving cognitive performance while at the same time decreasing Aβ production by an endogenously relevant mechanism. We propose that a sAPPα-enhancer would be effective as a monotherapy or as part of multi-modal therapy in AD. This enhancement may be induced by treatment with a variety of molecules as described in this review that increase sAPPα. Furthermore, sAPPα enhancement may also be an effective therapeutic approach in treatment of TBI, ALS, CAA, and stroke.
ACKNOWLEDGEMENTS
This work was supported by grants from the Mary S. Easton Center for Alzheimer’s Disease Research, Rosenberg Alzheimer’s Project, the Drown Foundation, Acceleration Partners, R21AG041456 from NIA, R01AG034427 from NIA, Bechtel Foundation, and Alzheimer’s Drug Discovery Foundation (ADDF).
REFERENCES
- Alzheimer’s Association (2015) 2015 Alzheimer's disease facts and figures. Alzheimers Dement 11: 332-384.
- Ikeda S, Wong CW, Allsop D, Landon M, Kidd M, et al. (1987) Immunogold labeling of cerebrovascular and neuritic plaque amyloid fibrils in Alzheimer’s disease with an anti-beta protein monoclonal antibody. Lab Invest 57: 446-449.
- Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: 383-388.
- Goedert M (1993) Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci 16: 460-465.
- Dickson DW (1997) Discovery of new lesions in neurodegenerative diseases with monoclonal antibody techniques: is there a non-amyloid precursor to senile plaques? Am J Pathol 151: 7-11.
- Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer’s disease. Neuron 44: 181-193.
- Devi L, Ohno M (2013) Effects of levetiracetam, an antiepileptic drug, on memory impairments associated with aging and Alzheimer’s disease in mice. Neurobiol Learn Mem 102: 7-11.
- Sanchez PE, Zhu L, Verret L, Vossel KA, Orr AG, et al. (2012) Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci USA 109: 2895-2903.
- Alagiakrishnan K, Sankaralingam S, Ghosh M, Mereu L, Senior P (2013) Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer’s disease. Discov Med 16: 277-286.
- Hölscher C (2014) First clinical data of the neuroprotective effects of nasal insulin application in patients with Alzheimer’s disease. Alzheimers Dement 10: 33-37.
- Eketjäll S, Janson J, Jeppsson F, Svanhagen A, Kolmodin K, et al. (2013) AZ-4217: a high potency BACE inhibitor displaying acute central efficacy in different in vivo models and reduced amyloid deposition in Tg2576 mice. J Neurosci 33: 10075-10084.
- May PC, Dean RA, Lowe SL, Martenyi F, Sheehan SM, et al. (2011) Robust central reduction of amyloid-β in humans with an orally available, non-peptidic β-secretase inhibitor. J Neurosci 31: 16507-16516.
- Wyss DF, Wang YS, Eaton HL, Strickland C, Voigt JH, et al. (2012) Combining NMR and X-ray crystallography in fragment-based drug discovery: discovery of highly potent and selective BACE-1 inhibitors. Top Curr Chem 317: 83-114.
- Descamps O, Spilman P, Zhang Q, Libeu CP, Poksay K, et al. (2013) AβPP-selective BACE inhibitors (ASBI): novel class of therapeutic agents for alzheimer’s disease. J Alzheimers Dis 37: 343-355.
- Glenner GG, Wong CW (1984) Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun 120: 885-890.
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, et al. (1985) Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 82: 4245-4249.
- Butterfield DA, Reed T, Newman SF, Sultana R (2007) Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med 43:658-677.
- Parameshwaran K, Dhanasekaran M, Suppiramaniam V (2008) Amyloid beta peptides and glutamatergic synaptic dysregulation. Exp Neurol 210: 7-13.
- Rosales-Corral S, Tan DX, Reiter RJ, Valdivia-Velázquez M, Acosta-Martínez JP, et al. (2004) Kinetics of the neuroinflammation-oxidative stress correlation in rat brain following the injection of fibrillar amyloid-beta onto the hippocampus in vivo. J Neuroimmunol 150: 20-28.
- Canevari L, Clark JB, Bates TE (1999) beta-Amyloid fragment 25-35 selectively decreases complex IV activity in isolated mitochondria. FEBS Lett 457: 131-134.
- Lin H, Bhatia R, Lal R (2001) Amyloid beta protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J 15: 2433-2444.
- Inestrosa NC, Alvarez A, Godoy J, Reyes A, De Ferrari GV (2000) Acetylcholinesterase-amyloid-beta-peptide interaction and Wnt signaling involvement in Abeta neurotoxicity. Acta Neurol Scand Suppl 176: 53-59.
- Galimberti D, Scarpini E (2010) Genetics and biology of Alzheimer’s disease and frontotemporal lobar degeneration. Int J Clin Exp Med 3: 129-143.
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, et al. (2012) A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 488: 96-99.
- Maloney JA, Bainbridge T, Gustafson A, Zhang S, Kyauk R, et al. (2014) Molecular mechanisms of Alzheimer disease protection by the A673T allele of amyloid precursor protein. J Biol Chem 289: 30990-31000.
- Sadigh-Eteghad S, Talebi M, Farhoudi M (2012) Association of apolipoprotein E epsilon 4 allele with sporadic late onset Alzheimer’s disease. A meta-analysis. Neurosciences (Riyadh) 17: 321-326.
- Holtzman DM, Herz J, Bu G (2012) Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med 2: 006312.
- Jack CR Jr, Knopman DS, Weigand SD, Wiste HJ, Vemuri P, et al. (2012) An operational approach to National Institute on Aging-Alzheimer’s Association criteria for preclinical Alzheimer disease. Ann Neurol 71: 765-775.
- Das U, Scott DA, Ganguly A, Koo EH, Tang Y, et al. (2013) Activity-induced convergence of APP and BACE-1 in acidic microdomains via an endocytosis-dependent pathway. Neuron 79: 447-460.
- Bekris LM, Lutz F, Li G, Galasko DR, Farlow MR, et al. (2012) ADAM10 expression and promoter haplotype in Alzheimer’s disease. Neurobiol Aging 33: 2229.
- Obregon D, Hou H, Deng J, Giunta B, Tian J, et al. (2012) Soluble amyloid precursor protein-α modulates β-secretase activity and amyloid-β generation. Nat Commun 3: 777.
- Peters-Libeu C, Jesus C, Mitsumori M, Poksay K, Spilman P, et al. (2015) sAßPPa is a potent endogenous inhibitor of BACE 1. J Alzheimer’s disease JAD Preprint: 1-11.
- Tian Y, Crump CJ, Li YM (2010) Dual role of alpha-secretase cleavage in the regulation of gamma-secretase activity for amyloid production. J Biol Chem 285: 32549-32556.
- Chasseigneaux S, Allinquant B (2012) Functions of Aβ, sAPPα and sAPPβ : similarities and differences. J Neurochem 120: 99-108.
- Ortega F, Stott J, Visser SA, Bendtsen C (2013) Interplay between α-, β-, and γ-secretases determines biphasic amyloid-β protein level in the presence of a γ-secretase inhibitor. J Biol Chem 288: 785-792.
- Gralle M, Botelho MG, Wouters FS (2009) Neuroprotective secreted amyloid precursor protein acts by disrupting amyloid precursor protein dimers. J Biol Chem 284: 15016-15025.
- Mattson MP, Furukawa K (1998) Signaling events regulating the neurodevelopmental triad. Glutamate and secreted forms of beta-amyloid precursor protein as examples. Perspect Dev Neurobiol 5: 337-352.
- Claasen AM, Guévremont D, Mason-Parker SE, Bourne K, Tate WP, et al. (2009) Secreted amyloid precursor protein-alpha upregulates synaptic protein synthesis by a protein kinase G-dependent mechanism. Neurosci Lett 460: 92-96.
- Anderson JJ, Holtz G, Baskin PP, Wang R, Mazzarelli L, et al. (1999) Reduced cerebrospinal fluid levels of alpha-secretase-cleaved amyloid precursor protein in aged rats: correlation with spatial memory deficits. Neuroscience 93: 1409-1420.
- Corrigan F, Vink R, Blumbergs PC, Masters CL, Cappai R, et al. (2012) Characterisation of the effect of knockout of the amyloid precursor protein on outcome following mild traumatic brain injury. Brain Res 1451: 87-99.
- Hick M, Herrmann U, Weyer SW, Mallm JP, Tschäpe JA, et al. (2015) Acute function of secreted amyloid precursor protein fragment APPsα in synaptic plasticity. Acta Neuropathol 129: 21-37.
- Almkvist O, Basun H, Wagner SL, Rowe BA, Wahlund LO, et al. (1997) Cerebrospinal fluid levels of alpha-secretase-cleaved soluble amyloid precursor protein mirror cognition in a Swedish family with Alzheimer disease and a gene mutation. Arch Neurol 54: 641-644.
- Suh J, Choi SH, Romano DM, Gannon MA, Lesinski AN, et al. (2013) ADAM10 missense mutations potentiate β-amyloid accumulation by impairing prodomain chaperone function. Neuron 80: 385-401.
- Kaden D, Harmeier A, Weise C, Munter LM, Althoff V, et al. (2012) Novel APP/Aβ mutation K16N produces highly toxic heteromeric Aβ oligomers. EMBO Mol Med 4: 647-659.
- Di Fede G, Catania M, Morbin M, Rossi G, Suardi S, et al. (2009) A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323: 1473-1477.
- Di Fede G, Catania M, Morbin M, Giaccone G, Moro ML, et al. (2012) Good gene, bad gene: new APP variant may be both. Prog Neurobiol 99: 281-292.
- Theendakara V, Patent A, Peters Libeu CA, Philpot B, Flores S, et al. (2013) Neuroprotective Sirtuin ratio reversed by ApoE4. Proc Natl Acad Sci USA 110: 18303-18308.
- Lattanzio F, Carboni L, Carretta D, Rimondini R, Candeletti S, et al. (2014) Human apolipoprotein E4 modulates the expression of Pin1, Sirtuin 1, and Presenilin 1 in brain regions of targeted replacement apoE mice. Neuroscience 256: 360-369.
- Rhinn H, Fujita R, Qiang L, Cheng R, Lee JH, et al. (2013) Integrative genomics identifies APOE ε4 effectors in Alzheimer’s disease. Nature 500: 45-50.
- Julien C, Tremblay C, Emond V, Lebbadi M, Salem N Jr, et al. (2009) Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol 68: 48-58.
- Kumar R, Chaterjee P, Sharma PK, Singh AK, Gupta A, et al. (2013) Sirtuin1: a promising serum protein marker for early detection of Alzheimer’s disease. PLoS One 8: 61560.
- Tippmann F, Hundt J, Schneider A, Endres K, Fahrenholz F (2009) Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J 23: 1643-1654.
- Donmez G, Wang D, Cohen DE, Guarente L (2010) SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell 142: 320-332.
- Donmez G (2013) Sirtuins as possible targets in neurodegenerative diseases. Curr Drug Targets 14: 644-647.
- Zhang Q, Descamps O, Hart MJ, Poksay KS, Spilman P, et al. (2014) Paradoxical effect of TrkA inhibition in Alzheimer’s disease models. J Alzheimers Dis 40: 605-617.
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, et al. (1999) Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA 96: 3228-3233.
- Wang W, Liu Y, Lazarus RA (2013) Allosteric inhibition of BACE1 by an exosite-binding antibody. Curr Opin Struct Biol 23: 797-805.
- Cumming JN, Smith EM, Wang L, Misiaszek J, Durkin J, et al. (2012) Structure based design of iminohydantoin BACE1 inhibitors: identification of an orally available, centrally active BACE1 inhibitor. Bioorg Med Chem Lett 22: 2444-2449.
- Spilman P, Descamps O, Gorostiza O, Peters-Libeu C, Poksay KS, et al. (2014) The multi-functional drug tropisetron binds APP and normalizes cognition in a murine Alzheimer’s model. Brain Res 1551: 25-44.
- Fiebich BL, Akundi RS, Lieb K, Candelario-Jalil E, Gmeiner D, et al. (2004) Antiinflammatory effects of 5-HT3 receptor antagonists in lipopolysaccharide-stimulated primary human monocytes. Scand J Rheumatol Suppl 119: 28-32.
- Setoguchi D, Nakamura M, Yatsuki H, Watanabe E, Tateishi Y, et al. (2011) Experimental examination of anti-inflammatory effects of a 5-HT3 receptor antagonist, tropisetron, and concomitant effects on autonomic nervous function in a rat sepsis model. Int Immunopharmacol 11: 2073-2078.
- Tasaka Y, Yasunaga D, Kiyoi T, Tanaka M, Tanaka A, et al. (2015) Involvement of stimulation of α7 nicotinic acetylcholine receptors in the suppressive effect of tropisetron on dextran sulfate sodium-induced colitis in mice. J Pharmacol Sci 127: 275-283.
- Meraz-Ríos MA, Toral-Rios D, Franco-Bocanegra D, Villeda-Hernández J, Campos-Peña V (2013) Inflammatory process in Alzheimer’s Disease. Front Integr Neurosci 7: 59.
- Lee HR, Shin HK, Park SY, Kim HY, Lee WS, et al. (2014) Cilostazol suppresses β-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-β. J Neurosci Res 92: 1581-1590.
- Endres K, Fahrenholz F, Lotz J, Hiemke C, Teipel S, et al. (2014) Increased CSF APPs-α levels in patients with Alzheimer disease treated with acitretin. Neurology 83: 1930-1935.
- Postina R (2012) Activation of α-secretase cleavage. J Neurochem 120: 46-54.
- Endres K, Fahrenholz F (2012) Regulation of α-secretase ADAM10 expression and activity. Exp Brain Res 217: 343-352.
- Qiu J, Li W, Feng SH, Wang M, He ZY (2014) Ginsenoside Rh2 promotes nonamyloidgenic cleavage of amyloid precursor protein via a cholesterol-dependent pathway. Genet Mol Res 13:3586-3598.
- Zhang SQ, Sawmiller D, Li S, Rezai-Zadeh K, Hou H, et al. (2013) Octyl gallate markedly promotes anti-amyloidogenic processing of APP through estrogen receptor-mediated ADAM10 activation. PLoS One 8: 71913.
- Lecoutey C, Hedou D, Freret T, Giannoni P, Gaven F, et al. (2014) Design of donecopride, a dual serotonin subtype 4 receptor agonist/acetylcholinesterase inhibitor with potential interest for Alzheimer’s disease treatment. Proc Natl Acad Sci USA 111: 3825-3830.
- Zhang SQ, Obregon D, Ehrhart J, Deng J, Tian J, et al. (2013) Baicalein reduces β-amyloid and promotes nonamyloidogenic amyloid precursor protein processing in an Alzheimer’s disease transgenic mouse model. J Neurosci Res 91: 1239-1246.
- Etcheberrigaray R, Tan M, Dewachter I, Kuipéri C, Van der Auwera I, et al. (2004) Therapeutic effects of PKC activators in Alzheimer’s disease transgenic mice. Proc Natl Acad Sci USA 101: 11141-11146.
- Auerbach ID, Vinters HV (2006) Effects of anoxia and hypoxia on amyloid precursor protein processing in cerebral microvascular smooth muscle cells. J Neuropathol Exp Neurol 65: 610-620.
- Bryson JB, Hobbs C, Parsons MJ, Bosch KD, Pandraud A, et al. (2012) Amyloid Precursor Protein (APP) contributes to pathology in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Hum Mol Genet 21: 3871-3882.
- Steinacker P, Fang L, Kuhle J, Petzold A, Tumani H, et al. (2011) Soluble beta-amyloid precursor protein is related to disease progression in amyotrophic lateral sclerosis. PLoS One 6: 23600.
- Yoon EJ, Park HJ, Kim GY, Cho HM, Choi JH, et al. (2009) Intracellular amyloid beta interacts with SOD1 and impairs the enzymatic activity of SOD1: implications for the pathogenesis of amyotrophic lateral sclerosis. Exp Mol Med 41: 611-617.
- Valentine JS, Doucette PA, Zittin Potter S (2005) Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem 74: 563-593.
- Vehviläinen P, Koistinaho J, Gundars G (2014) Mechanisms of mutant SOD1 induced mitochondrial toxicity in amyotrophic lateral sclerosis. Front Cell Neurosci 8: 126.
- Li QX, Mok SS, Laughton KM, McLean CA, Volitakis I, et al. (2006) Overexpression of Abeta is associated with acceleration of onset of motor impairment and superoxide dismutase 1 aggregation in an amyotrophic lateral sclerosis mouse model. Aging Cell 5:153-165.
- Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW, Moussa CE (2011) β-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res 1386: 191-199.
- Combes M, Poindron P, Callizot N (2015) Glutamate protects neuromuscular junctions from deleterious effects of β-amyloid peptide and conversely: an in vitro study in a nerve-muscle coculture. J Neurosci Res 93: 633-643.
- Rabinovich-Toidman P, Becker M, Barbiro B, Solomon B (2012) Inhibition of amyloid precursor protein beta-secretase cleavage site affects survival and motor functions of amyotrophic lateral sclerosis transgenic mice. Neurodegener Dis 10: 30-33.
- Watanabe S, Ageta-Ishihara N, Nagatsu S, Takao K, Komine O, et al. (2014) SIRT1 overexpression ameliorates a mouse model of SOD1-linked amyotrophic lateral sclerosis via HSF1/HSP70i chaperone system. Mol Brain 7: 62.
- Song L, Liang C, Zhang X, Li J, Le W (2014) Resveratrol ameliorates motor neuron degeneration and improves survival in SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Biomed Res Int 483501.
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, et al. (2007) SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 26: 3169-3179.
- Dombovy ML (2011) Traumatic brain injury. Continuum (Minneap Minn) 17: 584-605.
- Yang ST, Hsiao IT, Hsieh CJ, Chiang YH, Yen TC, et al. (2015) Accumulation of amyloid in cognitive impairment after mild traumatic brain injury. J Neurol Sci 349: 99-104.
- Shively S, Scher AI, Perl DP, Diaz-Arrastia R (2012) Dementia resulting from traumatic brain injury: what is the pathology? Arch Neurol 69: 1245-1251.
- Olsson A, Csajbok L, Ost M, Höglund K, Nylén K, et al. (2004) Marked increase of beta-amyloid(1-42) and amyloid precursor protein in ventricular cerebrospinal fluid after severe traumatic brain injury. J Neurol 251: 870-876.
- Mannix RC, Zhang J, Berglass J, Qui J, Whalen MJ (2013) Beneficial effect of amyloid beta after controlled cortical impact. Brain Inj 27: 743-748.
- Mannix RC, Zhang J, Park J, Lee C, Whalen MJ (2011) Detrimental effect of genetic inhibition of B-site APP-cleaving enzyme 1 on functional outcome after controlled cortical impact in young adult mice. J Neurotrauma 28: 1855-1861.
- Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, et al. (2010) The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One 5: 9505.
- Thornton E, Vink R, Blumbergs PC, Van Den Heuvel C (2006) Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res 1094: 38-46.
- Moussavi Nik SH, Wilson L, Newman M, Croft K, Mori TA, et al. (2012) The BACE1-PSEN-AβPP regulatory axis has an ancient role in response to low oxygen/oxidative stress. J Alzheimers Dis 28: 515-530.
- Zhang X, Zhou K, Wang R, Cui J, Lipton SA, et al. (2007) Hypoxia-Inducible Factor 1alpha (HIF-1alpha)-mediated hypoxia increases BACE1 expression and beta-amyloid generation. J Biol Chem 282: 10873-10880.
- Nalivaeva NN, Turner AJ (2013) The amyloid precursor protein: a biochemical enigma in brain development, function and disease. FEBS Lett 587: 2046-2054.
- Li QY, Wang HM, Wang ZQ, Ma JF, Ding JQ, et al. (2010) Salidroside attenuates hypoxia-induced abnormal processing of amyloid precursor protein by decreasing BACE1 expression in SH-SY5Y cells. Neurosci Lett 481: 154-158.
- Siopi E, Llufriu-Dabén G, Cho AH, Vidal-Lletjós S, Plotkine M, et al. (2013) Etazolate, an α-secretase activator, reduces neuroinflammation and offers persistent neuroprotection following traumatic brain injury in mice. Neuropharmacology 67: 183-192.
- Agostinho P, Pliássova A, Oliveira CR, Cunha RA (2015) Localization and Trafficking of Amyloid-β Protein Precursor and Secretases: Impact on Alzheimer’s Disease. J Alzheimers Dis 45: 329-347.
- Musiek ES, Xiong DD, Holtzman DM (2015) Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp Mol Med 47: 148.
- Ozcankaya R, Delibas N (2002) Malondialdehyde, superoxide dismutase, melatonin, iron, copper, and zinc blood concentrations in patients with Alzheimer disease: cross-sectional study. Croat Med J 43: 28-32.
- Ferrari E, Arcaini A, Gornati R, Pelanconi L, Cravello L, et al. (2000) Pineal and pituitary-adrenocortical function in physiological aging and in senile dementia. Exp Gerontol 35: 1239-1250.
- Olcese JM, Cao C, Mori T, Mamcarz MB, Maxwell A, et al. (2009) Protection against cognitive deficits and markers of neurodegeneration by long-term oral administration of melatonin in a transgenic model of Alzheimer disease. J Pineal Res 47: 82-96.
- Shukla M, Htoo HH, Wintachai P, Hernandez JF, Dubois C, et al. (2015) Melatonin stimulates the nonamyloidogenic processing of βAPP through the positive transcriptional regulation of ADAM10 and ADAM17. J Pineal Res 58: 151-165.
Citation: Spilman P, Bredesen DE, Jagodzinska B, John V (2015) Enhancement of sAPPα as a Therapeutic Strategy for Alzheimer’s and Other Neurodegenerative Diseases. J Alzheimers Neurodegener Dis 1: 001.
Copyright: © 2015 Patricia Spilman, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.