HOX Genes and Cancer Stem Cells: Update on HOX expression, DNA Methylation, Biomarker Status, and Genetic Changes in Colorectal Cancer
*Corresponding Author(s):
Bruce M BomanThomas Jefferson University, United States
Tel:+1 2673039241,
Email:brboman@udel.edu
Abstract
HOX genes are best known for their ability to control embryonic Stem Cells (SCs) that specify body pattern segmentation during development. HOX genes also regulate adult SCs by specifying SC differentiation into specialized cell lineages. Moreover, in most human cancers, HOX genes are aberrantly expressed; but the mechanisms that explain how HOX genes contribute to cancer etiology need to be clarified. Our goal herein is to review current research findings on HOX genes in SCs in cancers, and to provide an update from our bioinformatics analysis of HOX genes in Colorectal Cancer (CRC). The updated results show that most HOX genes are overexpressed in CRC. Moreover, select HOX genes are hyper-methylated and the pattern of hyper-methylation is associated with APC mutation. Additionally, aberrant expression of many HOX genes (22 of 39) predicts (p<0.05) decreased survival of CRC patients. Continued discovery of the mechanisms that explain how HOX genes regulate normal colon SCs and how dysregulation of HOX genes in cancer SCs drive CRC growth should provide insight into strategies for development of new SC-targeted therapies for CRC.
Keywords
Adenomatous polyposis coli; Colon cancer; DNA methylation; HOX genes, Stem cells
INTRODUCTION
HOX genes are a set of highly conserved genes that dictate the general body plan of multicellular organisms. In mammals, 39 HOX genes are categorized into four groups or gene clusters (A-D), with each group found on a different chromosome. Each gene within a group is also assigned a number. In humans, there are 13 paralogous groups containing 9 to 11 members. HOX genes that share a number are paralogous to each other. For example, HOXA1 and HOXA4 are on the same chromosome, but HOXA1 and HOXB1 are paralogous to each other. HOX genes are comprised of two exons and one intron, with the latter exon containing the codon sequence for the homeobox domain which encodes a HOX protein [1]. One unique trait of HOX gene groups is that genes toward the 3’ end are typically expressed towards the anterior of the organism, while those at the 5’ end are expressed towards the posterior. In this way, the diversely regulated repertoire of HOX genes serves to specify tempero-spatial collinearity in anterior-posterior axial patterning [2]. HOX proteins serve as master transcriptional factors that regulate Stem Cell (SC) differentiation in embryonic development and homeostatic SC processes in adults. Dysregulation of HOX gene function thereby disrupts normal SC homeostasis and promotes tumorigenesis [1,3,4].
Stem cells are undifferentiated cells that are capable of self-renewal and differentiating into different cell types. SC fate is determined by developmental regulatory signals and niche-specific environmental factors including WNT, Hedgehog, Notch, BMP, fibroblast and epidermal growth factors [5]. Normal SCs can be categorized into three general types: Embryonic (ESCs), Adult (ASCs), and induced Pluripotent SCs (iPSCs) [1]. As their name suggests, ESCs are found within the blastocysts of embryos. These cells are known to be pluripotent, meaning they can differentiate into all three germ cell types (ectoderm, mesoderm, and endoderm). ASCs, also known as “tissue-specific SCs”, are found throughout the body and typically differentiate into a small group of specialized cell types. In breast tissue for instance, ASCs can be found within Terminal Ductal Lobuloalveolar Units (TDLUs) [6]. Lastly, iPSCs are pluripotent SCs artificially generated from somatic cells [7]. Due to the self-renewing property of SCs, dysregulation of SC function can result in the formation of a fourth type: cancer SCs (CSCs) [1]. CSCs, like normal SCs, are undifferentiated cells with the ability to self-renew and differentiate. CSCs are vital in promoting tumor growth and are common initiators of cancer metastasis [3]. CSCs also confer resistance to radiation and chemotherapy, which can be exacerbated by dysregulation of certain HOX genes such as HOXB7 [3] and HOXB13 [8]. With the identity of the following cell markers to distinguish CSCs from other cell types: CD24, CD29, CD44, CD90, CD133 and CD166 [9], targeting CSCs may prove significant in treating cancers.
HOX GENE REGULATION, EXPRESSION, AND FUNCTION IN NORMAL STEM CELLS
Embryonic Stem Cells (ESCs)
HOX genes typically are not transcribed in early ESCs. In fact, HOX genes have been shown to be epigenetically repressed in ESCs until the embryo undergoes gastrulation [3]. The suppression of HOX clusters allows HOX gene expression to subsequently become activated as ESCs commit to distinct lineages. Several factors undo this HOX gene repression, such as histone demethylases. Also, WNT and Fibroblast Growth Factor (FGF) versus Retinoic Acid (RA) signaling pathways have opposing effects on HOX gene expression that controls ESC differentiation and axial pattern formation during formation of the vertebrate embryo body [10]. Many properties of ESCs with regard to HOX gene expression also apply to iPSCs, which provides a means to study HOX gene regulation [9]. The temporal and spatial mechanisms by which expression of the transcription factors encoded by HOX genes regulate ESCs to establish precise tissue patterning during morphogenesis in the vertebrate embryo remain a focus of intense research in developmental biology. Nevertheless, the growing body of scientific evidence indicates it is the pattern of HOX expression, rather than any one of the individual HOX genes, that regulates the behavior of ESCs during embryogenesis.
Because HOX genes play such a critical role during development, their loss or gain of function can lead to the homeotic transformation and abnormal formation of body tissues. The necessity to maintain proper formation of organized tissue structure and body plan explains why the basic structure of HOX genes has been evolutionarily conserved in metazoan animals [11]. As transcription factors, different expression patterns of the 39 HOX proteins are intricately maintained during embryogenesis as a coordinated effort of multiple cellular and developmental pathways [10]. The synthesis of HOX proteins begins with transcription of its genes in a complicated and intricate manner. One way transcription is initiated and regulated is through the binding of RA to RA receptors which complex with RA Responsive Elements (RAREs) located within the vicinity of HOX genes. Indeed, several HOX genes are regulated via RAREs located within their HOX gene cluster [10]. The inhibition of HOX transcription occurs by the absence of RA binding to Retinoic Acid Receptor (RAR) and Retinoid X Receptor (RXR) heterodimers which occupy the RAREs, as well as the involvement of other suppressive regulatory factors [12-15]. In the event an RA is bound to the RAR-RXR heterodimer, an allosteric change of the complex releases the suppressive regulatory factors, and allows recruitment of co-activators to stimulate transcription [12-14,16]. One critical component of the RA-HOX gene interaction is RA Receptor γ (RARγ), which is required for HOX gene transcription in ESCs. Deletion of RARγ has been shown to severely reduce the efficacy of RA in inducing HOX gene expression [3].
Studies exploring the mechanisms of RA signaling in terms of targeting specific Hox paralogues and individual Hox genes have revealed their role in tempero-spatial collinearity. For example, Hox genes closer to the 3’-end of the chromosome, specifically Hoxa1, Hoxb1, and Hoxd1, are much more sensitive to fluctuation in RA concentration. This coincides with the fact that RAREs are generally located closer to the 3’-end of the chromosome rather than the 5’-end [17,18]. However, some RAREs have been shown to regulate Hox expression more globally over longer ranges [19]. The temporal difference between activation of Hoxa1 and Hoxb1 in mouse embryos is attributed to differential RA response mechanisms. The time delay of Hoxb1 activation, compared to Hoxa1 activation, may be due to Hoxa1 being constitutively suppressed in the absence of RA and bound to an RAR-RXR complex. However, Hoxb1 actively recruits RAR-RXR in the presence of retinoid acid ligands, and thus stimulates its own transcription. It was reported that the rapid activation of Hoxa1 might be due to the release of suppression (a second order reaction), which is kinetically faster than a de novo recruitment of RA as well as RAR-RXR (close to a 3rd order reaction), in the case of Hoxb1 activation. Similar mechanisms have been widely observed in ESCs [20,21].
The studies we discussed above show that RA signaling is a direct regulatory mechanism for Hox expression, which logically establish the fact that all upstream pathways that cause fluctuations in RA availability (e.g. RA synthesis and degradation) would virtually affect Hox expression as well. It has been reported in multiple animal models that the majority of biological RA synthesis is catalyzed by Aldh1a2 [22-25]. During embryogenesis, FGF signaling is another major pathway that represses RA signaling. In mammalian embryos, FGF, together with Wnt, are important in maintaining a posterior zone rich in stem cells (sometimes referred to as caudal stem zone). In this zone, FGF and Wnt have been reported to oppose RA signaling possibly via inhibiting the expression of Aldh1a2, but also being reciprocally repressed by RA, as the product of Aldh1a2, in other parts of the embryo [26-33]. While in the more anterior region (midbrain and hindbrain), FGF has been reported to instigate the expression of Cyp26, which catalyzes RA degradation [27,34-36]. Based on the interweaved complexity established by the signaling pathways, the precise control of the modularized HOX expression domain must be highly conserved [17] to insure proper differentiation and tissue homeostasis during animal development [37].
Adult Stem Cells (ASCs)
The expression of HOX genes in ASCs is heavily dependent on the particular tissue type. In the adult, HOX genes play a crucial role in the function of ASCs by specifying differentiation into specialized cell lineages, maintaining cell organization and governing homeostasis [3]. For example, HOXA4, HOXA9, and HOXD10 have all been shown to regulate colonic SC self-renewal and proliferation. More specifically, they are selectively expressed within the colonic SCs at the bottom of the human colonic crypt, not in the differentiated daughter cells located at the crypt top [1]. In particular, HOXA4 and HOXA9 are linked to crypt cell proliferation while HOXC8 and HOXC9 are linked to crypt cell differentiation [5]. Hematopoietic SCs, meanwhile, are closely linked to HOXB3, HOXB4 and HOXA9 [1]. For example, HOXB4 is a key regulator of hematopoietic SC differentiation, and regulates transcription factors for several genes related to hematopoiesis, while HOXB3 has been linked to the differentiation of B and T cells [3]. HOXB3/HOXB4 double knockout mice have been shown to have severely stunted hematopoiesis [3]. HOXA9 is one of the most active HOX genes in hematopoietic SC, and is linked with cell proliferation. The reader is referred to reference [1] for more detailed information on role of HOX genes in ASCs, including mesenchymal ASCs. The above findings indicate that HOX genes play diverse roles in normal SC functions and properties, from self-renewal to multi-lineage differentiation. Therefore, our understanding of the molecular mechanisms for how HOX genes control SC self-renewal and differentiation will ultimately help us understand how SC populations are maintained in normal, disease-free states and how the dysregulation of HOX genes contributes to cancer development through aberrant self-renewal and differentiation of SCs.
ABERRANT HOX GENE EXPRESSION, EMERGENCE OF CANCER STEM CELLS AND TUMOR DEVELOPMENT
Due to their role in regulating SC differentiation and proliferation, dysregulation of several HOX genes has been linked with carcinogenesis. Typically, tumor growth occurs when overexpression or repression of a gene alters a cell’s ability to differentiate. For instance, overexpression of HOXA9, a gene associated with cell proliferation, has been linked with both Colorectal Cancer (CRC) [5,38,39] and leukemia [1]. HOXA4 is another cell proliferation HOX gene associated with CRC [1,5]. It is also interesting to note that the level of dysregulation for a particular HOX gene not only changes based on the type of cancer, but on the stage as well. Evidence of this is seen in CRC, where HOX overexpression in stage II was shown to decrease in stage III, only to go back up in stage IV [38,39].
Loss of function of HOXA5 in breast cancer results in the loss of epithelial traits such as intercellular adhesion [40]. Furthermore, cells that lose proper function of HOXA5 have reduced protein expression of CDH1, CD24 and p53, all three of which play a role in apoptosis [41]. Reintroduction of HOXA5 causes these under-expressed proteins to regain normal expression patterns [40]. It is suspected that HOXA5 plays a role in the RA pathway [9]. HOXA5 has also been shown to play a similar role in cervical cancer, with most cervical cancers having a significant down regulation of the gene [42]. When HOXA5 is properly expressed in cervical cells, it has a proliferation inhibiting effect caused by the promotion of p53 and inhibition of the WNT/β-catenin pathway [42].
Improper HOXA9 expression has been linked to a myriad of cancers, including CRC [5] and well-documented in acute myeloid leukemia [3]. In most leukemia cases, HOXA9 is overexpressed and acts as a transcription factor for multiple oncogenes [43] such as NUP98 [44]. Furthermore, HOXA9 overexpression strongly correlates with poor prognosis [44]. Due to this well-established link, several chemical agents targeting HOXA9 have been developed. HXR9 peptide inhibitor targets HOXA9/PBX protein complexes [44] and heterocyclic diamidine DNA ligands [43] are designed to prevent HOXA9 from binding to the DNA. While links between HOXA9 overexpression and CRC have been observed [5], their mechanisms are not as well understood as compared to leukemia.
Overexpression of HOXB3 promotes tumor growth in a myriad of ways. HOXB3 binds to and activates the methyltransferase DNMT3B, which in turn silences the tumor suppressor gene RASSF1A via methylation [45]. As such, HOXB3 overexpression causes downregulation of RASSF1A resulting in tumor growth. Another result of HOXB3 overexpression is tamoxifen resistance [45]. Tamoxifen is a commonly used breast cancer treatment drug that targets Estrogen Receptor-α (ERα). HOXB3 also promotes expression of CD44, a cell surface protein that positively correlates with CSC phenotypic expression. HOXB3 expression is reduced by the introduction of microRNA-375 (miR-375) via RNA interference (RNAi) [46]. As such, introducing miR-375 to MCF-7 CSCs results in susceptibility to tamoxifen and a reduction of CD44 and other CSC traits.
HOXB8 is another HOX gene shown to influence tumor progression when it is improperly regulated. More specifically, in CRC cells, the upregulation of HOXB8 promotes cell proliferation. Furthermore, this proliferation is inhibited by HOXB8 knockdown resulting in apoptosis. One of the mechanisms by which these processes occur is via the WNT/β-catenin signaling pathway [47]. Another mechanism by which HOXB8 regulates cell growth is through the activation of Epithelial-Mesenchymal Transition (EMT) and Signal Transducer and Activator of Transcription 3 (STAT3) [48].
HOXB13 shares several traits with HOXB3. Much like HOXB3, HOXB13 is often overexpressed in breast cancer with HOXB13 expression being a poor prognosis for the disease [48]. Overexpression of HOXB13 confers tamoxifen resistance in breast CSCs. HOXB13 achieves this effect by directly repressing ERα expression. The oncoprotein Hepatitis B X-Interacting Protein (HBXIP) binds to and acetylates HOXB13, which severely reduces tamoxifen resistance [8]. Furthermore, HOXB13 promotes tumor formation via interleukin-6 (IL-6) expression, which plays a role in the mTOR pathway [49]. This pathway regulates cell proliferation and fibroblast recruitment. While not directly related to breast cancer, several HOXB13 SNP mutations have been linked to other types of cancer. For example, the G84E, Y88D, and L144P are all SNP mutations commonly found in prostate cancer [50].
HOXC8 is another HOX gene implicated in breast cancer development. Specifically, HOXC8 promotes transcription of intercellular adhesion protein Cadherin-11 (CDH11) [51]. Overexpression of HOXC8 causes upregulated transcription of CDH11, which leads to tumorigenesis. Inhibited expression of Embigin (EMB) by HOXC8, also contributes to tumorigenesis [52]. HOXC8 overexpression is also found in Esophageal Squamous Cell Cancer (ESCC), where overexpression is alleviated by long noncoding RNA- SNHG1 [9]. HOXC8 does not only contribute to breast cancer development when overexpressed; similar to HOXA5, when HOXC8 is downregulated in breast SCs, differentiation is inhibited, leading to tumorigenesis [53]. This is the result of methylation of the miR-196 promoter. Re-expression of HOXC8 is shown to reverse this effect. While this study identifies downregulation of HOXC8 by methylation of miR-196, the exact mechanism that causes tumorigenesis is not as well understood.
HOXD10, similarly to HOXA9 and HOXA4, has been linked to CRC [38,39], but with a few key differences. One of the most notable differences is that unlike HOXA9, HOXD10 is usually downregulated in CRC cells. Under normal conditions, HOXD10 promotes apoptosis by inhibiting the RHOC/AKT/MAPK signaling pathway. However, inhibition is reversed by hyper-methylation of the HOXD10 gene, resulting in CRC [43]. Decreasing the methylation reverses the apoptotic inhibitory effect.
EPIGENETIC REGULATION OF HOX GENES: DNA METHYLATION
DNA methylation is one of the most commonly studied epigenetic regulatory mechanisms. In DNA methylation, methyl groups locate to carbon 5 of cytosine nucleotides (m5C) that precede guanine nucleotides (commonly referred to as CpG islands) to alter transcriptional activity of DNA segments in their vicinity [54]. A 1990 model published by Fearon and Vogelstein hypothesized that carcinogenesis in CRC may be attributed to the accumulation of genetic alterations [55]. These authors briefly reviewed methylation patterns in colorectal tumors and proposed a theory that epigenetic alterations, such as loss and gain of DNA methylation, contribute to genomic instability and carcinogenesis [55]. This theory later led to discoveries which identified altered methylation of promoter regions as a major epigenetic mechanism of CRC. This type of alteration constitutes approximately 15-20% of all CRC cases and is collectively known as CpG Island Methylator Phenotype (CIMP) [56-58].
HOX genes encode a repertoire of transcription factors that either activate or repress specific gene targets that control proliferation, differentiation, migration, apoptosis and other hallmarks of cancer [59]. HOX transcription factors are further regulated by DNA methylation, and its application as a therapeutic target is recently discussed [60]. We previously reported that HOX genes are extensively involved in SC differentiation and carcinogenesis [1,61]. Altogether, this raises the possibility that specific HOX gene methylation patterns occur in different cancer types. We present bioinformatics data including methylation of HOX genes in CRC patients (see below).
It is advised to keep in mind however, the complicated nature of investigating DNA methylation patterns. Since DNA methylation exerts its function by regulating gene transcription, evaluation of its significance also depends on the level of specific mRNA present in the cell. Determining the amount of mRNA transcribed is further inherently confounded by other regulatory mechanisms such as that caused by long non-coding RNAs (lncRNAs). LncRNA HOTAIR for instance, regulates the expression of HOX genes presumably by modulating its chromatin state. HOTAIR is shown to be overexpressed in CRC [62]. This renders interpretation of methylation data rather difficult, and often dependent upon examination and verification of additional variables such as the corresponding gene expression level.
A genome-wide methylation study identified seven HOX genes (HOXB3, HOXB4, HOXB6, HOXB7, HOXC4, HOXC5, and HOXC6) that are significantly differentially methylated at transcription start sites in epithelial cell samples collected from the proximal and distal region of the colon [63]. Since the proximal and distal regions of the colon are of different embryonic origins (midgut and hindgut, respectively) [64], differential methylation could be directly related to the function of HOX genes in directing tissue positioning during development. In addition, the observation that polyps and cancers more frequently occur on the left side of the colon may be attributed to differential methylation of HOX genes [65]. While the pathologies of left-sided (distal) and right-sided (proximal) CRC have been extensively discussed at the genetic level [64-66], further investigation into differential HOX expression and methylation in particular would be very interesting.
Another group of abnormally methylated HOX genes (HOXA3, HOXC9, HOXC10, HOXC11 and HOXC13) were found in Aberrant Crypt Foci (ACF) [67]. ACFs are considered by many to be precursors to adenomas or polyps that if left untreated might develop into colorectal tumors [67]. Subject to the limitations of their corresponding methodologies (see further discussion below), all of these genes have been reported to be upregulated in CRC [1,61,68].
In terms of prognostic markers and patient survival, HOXC6 was recently implicated. The expression of HOXC6 was associated with metastasis and negatively correlated with patient survival expectancy in CRC [69]. Another study named HOXC6 as part of a “4-gene prognostic signature”, the expression levels of which can predict CRC patient survival [70]. Being that HOXC6 is relevant to CRC prognosis and that it is significantly more highly methylated on the right side of colon [63], it is not surprising that the expression level of HOXC6 is significantly higher in CIMP patients compared to non-CIMP patients [71].
STUDYING HOX GENE MECHANISMS IN NORMAL AND MALIGNANT HUMAN COLONIC STEM CELLS
We have been investigating HOX mechanisms in SCs for several reasons. 1) Embryonic SCs are controlled by HOX genes which specify tissue organization and body pattern segmentation during development [72-74]. 2) HOX genes also regulate adult SCs by specifying SC differentiation into specialized cell lineages. 3) HOX genes are aberrantly expressed in most human cancers [3,50,61,75,76]; but the role of HOX genes in cancer etiology is severely under-studied [77]. This is puzzling as other developmental regulatory pathways (WNT, Hedgehog, Notch, BMP) involved in embryo pattern segmentation play major roles in cancer etiology and have been extensively studied [78]. Perhaps HOX genes are less-well-investigated due to their idiosyncratic properties of function, structure and expression in any given tissue. Indeed, HOX genes encode master regulatory transcription factors (TFs) that regulate the maintenance of SC populations and their maturation into cell lineages. The complex TF regulatory network encoded by the 39 HOX genes acts like computer “motherboard” circuitry in SCs. That is, the HOX gene network coordinates connections between many of the crucial signaling components that maintain stemness of SCs and that regulate their differentiation. This coordination explains why expression of key HOX genes affects SC plasticity and why HOX genes are recognized as “master regulators”. For example, HOX genes are critical to adult neural SC and hematopoietic SC fate specification and differentiation into specialized cell lineages [1,79-86]. Moreover, the overexpression of HOX genes in leukemias is linked to blocked maturation of leukemic SCs at defined stages of maturation.
For the past decade, we have focused on the role of HOX genes in the SC overpopulation that drives CRC development. Initially, we showed by gene profiling and microdissection of human colon crypts that HOXA4 and HOXD10 are selectively expressed in human colon SCs and that HOXC8 and HOXC9 are expressed in differentiated colon cells [38]. qPCR and immunostaining confirmed co-localization of both HOXA4 and HOXD10 genes with SC markers ALDH1 and CD166 within the SC niche. We then studied how these HOX genes promote human CRC growth using immunohistochemical mapping, co-expression with several SC markers (ALDH1, CD166, LGR5, LRIG1), organoid formation, siRNA knockdown, TF regulatory network analysis, and bioinformatics analyses [1,5,38,39,41]. For example, results from our Promoter Analysis and Interaction Network Tool (PAINT) analysis identified differences in Transcription Factors (TFs) that regulate HOX genes in normal colon versus CRC [39]. This suggests that different HOX genes are controlled by different TFs, and the findings begin to elucidate how these HOX genes are dysregulated in CRC.
We further studied HOX expression in Aldehyde Dehydrogenase positive (ALDH+) colon SCs because ALDH is a major component of the Retinoic Acid (RA) signaling pathway and retinoids directly regulate many HOX genes through RAREs in their cluster sequence [87]. We found that HOXA4 and HOXD10 along with ALDH and the other components of the RA signaling pathway are selectively expressed in colon SCs [5,38,88,89]. Moreover, siRNA knockdown of HOXA4 and HOXD10 gene expression reduced ALDH+ SC numbers, cell proliferation, SC self-renewal and sphere formation [5]. Therefore, our findings indicate that RA regulation of HOX gene expression mainly occurs through ALDH+ SCs in the colon.
Elucidating how HOX genes might be involved in RA signaling regulation of differentiation of ALDH+ SC along the Neuroendocrine Cell (NEC) lineage has also been under our investigation [90]. Our rationale is as follows: 1) NEC populations occur throughout the GI tract and are a major part of the enteric nervous system [91-96]. 2) Retinoids induce differentiation of SCs into neural cell subtypes by regulating HOX expression [79,80]. Indeed, we found that All-Trans Retinoic Acid (ATRA) treatment of CRC cells reduces HOXA4 and HOXD10 expression and increases differentiation of SCs along the NEC lineage [88,90]. Thus, our findings suggest that retinoids regulate differentiation of colon SCs along the NEC lineage by controlling HOX genes.
We extended our study of HOX genes herein by using bioinformatics to analyze overexpression, hyper-methylation, mutation, and copy number gain of HOX genes in CRCs, and identify HOX genes that predict CRC patient survival. We found that most HOX genes are overexpressed in human CRCs and a subset (~30%) of HOX genes show some degree of hyper-methylation (Figure 1). In addition, we also found that aberrant expression of several HOX genes (22 of 39) predicts (p<0.05) decreased survival of CRC patients (Figure 2).We also found that the pattern of hyper-methylation of select HOX genes is associated with APC mutation (Table 1, Figure 3 and Supplemental Materials). These updated findings provide insight into the complexity of HOX gene expression and how HOX expression, when dysregulated, contributes to the development of CRC.
SUMMARY
Our goal herein was to review current research findings on the 39 member HOX gene family in normal SCs and CSCs, and to provide an update from our bioinformatics analysis of HOX genes in CRC. To explain our findings derived from bioinformatics analyses, we surmise that the HOX gene network might act like a computer motherboard “circuitry” in SCs (Figure 4). In this view, the HOX gene network processes and communicates information to other cellular processes that determine tissue phenotype. In embryogenic development, WNT, RA, and FGF signaling regulates HOX gene expression (reviewed in [10]). The pattern of HOX gene expression, in turn, controls the expression of HOX target genes, which specifies body pattern segmentation and underlies organ development. In this view, the function of the HOX “circuitry” is variable and can depend on DNA methylation via genomic imprinting as well as spatial and temporal feedback mechanisms occurring during head-to-tail tissue formation in embryogenesis.
In adult tissues, the HOX gene network in stem cells continues to be controlled by WNT, RA, FGF, and other signaling pathways. However, the function of the HOX “circuitry” may differ in each of our various bodily tissues. In this way, the specific HOX gene expression patterns would differ from tissue to tissue. And, differences in HOX expression would lead to regulation of a different set of HOX target genes, which would control tissue phenotype by specifying SC differentiation into specialized cell lineages, maintaining cell organization and governing homeostasis.
In cancer tissues, an imbalance in RA, WNT, FGF, or other signaling pathways might lead to dysregulation of HOX gene expression in malignant stem cells. Indeed, APC mutations occur in about 80% of CRCs and other WNT driver mutations (e.g. beta-catenin) also occur. Taken together, these mutations lead to activation of WNT signaling in about 95% of all CRCs. We found that sequential inactivation of APC in Familial Adenomatous Polyposis (FAP) patient colon tissues leads to progressive increase in the size of the ALDH+ SC population [89]. Since ALDH is a major component of the RA signaling pathway and retinoids directly regulate many HOX genes [87], we investigated HOX gene expression in ALDH+ SCs. We found that HOXA4 and HOXD10 along with ALDH and the other components of the RA signaling pathway are selectively expressed in colon SCs [5,38,88,89]. Moreover, siRNA knockdown of HOXA4 and HOXD10 gene expression reduced ALDH+ SC numbers, cell proliferation, SC self-renewal and sphere formation [5]. Therefore, our findings indicate that RA regulation of HOX gene expression mainly occurs through ALDH+ SCs in the colon and that APC mutations lead to attenuated RA signaling in ALDH+ SCs during CRC development. Since APC mutation causes increased WNT signaling, this suggests that increased WNT and decreased RA signaling in CRC lead to aberrant HOX gene expression.
Indeed, the activation of WNT signaling in majority of CRCs may explain why we found that the expression of most HOX genes is altered in CRCs (Figure 1A). The function of the HOX “circuitry” may also become altered by HOX mutations, changes in DNA methylation and other epigenetic modifications. Indeed, we found that 30% of the 39 HOX genes have some degree of DNA hyper-methylation (Figure 1B). We also found that the pattern of hyper-methylation of select HOX genes is associated with APC mutation (Table 1, Figure 3 and Supplemental Materials). Such alterations would lead to aberrant HOX expression patterns and dysregulation of target gene expression. Ultimately, HOX gene dysregulation in cancer would lead to CSC overpopulation, decreased cell differentiation, and disorganization of cells in tumor tissues. The outcome of these changes could affect the biological behavior of CRCs. Indeed, we found that aberrant expression of many HOX genes predicts decreased survival of CRC patients (Figure 2).
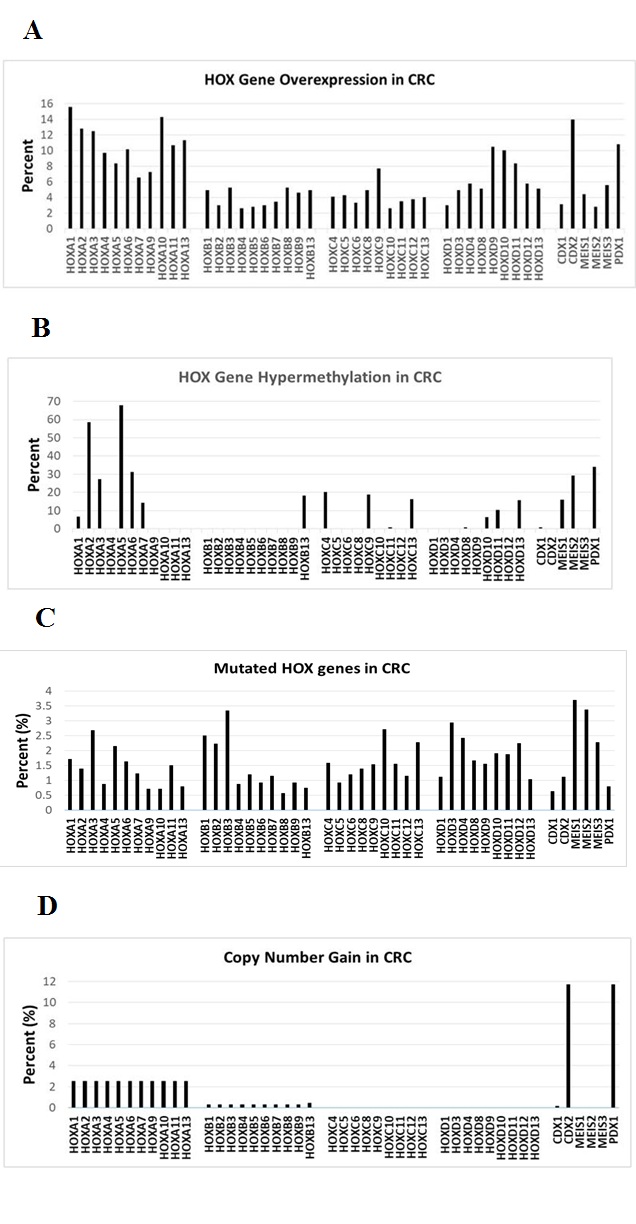 Figure 1: Bioinformatic analyses of HOX genes in CRC, including overexpression (A), hyper-methylation (B), mutations (C), copy number gain (D). Bioinformatics data derived from Cosmic Catalogue of Somatic Mutations in Cancer (https://cancer.sanger.ac.uk/cosmic).
Figure 1: Bioinformatic analyses of HOX genes in CRC, including overexpression (A), hyper-methylation (B), mutations (C), copy number gain (D). Bioinformatics data derived from Cosmic Catalogue of Somatic Mutations in Cancer (https://cancer.sanger.ac.uk/cosmic).
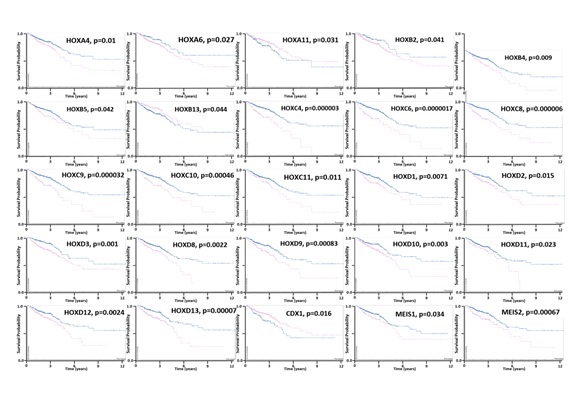 Figure 2: Kaplan-Meier survival analysis of HOX genes that predict (p < 0.05) patient survival. Curves reflect low (blue) and high (red) HOX gene expression. Y axis = Survival probability (0.0-1.0). X axis = Time (years 0-12). Bioinformatics data derived from The Human Protein Atlas (https://www.proteinatlas.org).
Figure 2: Kaplan-Meier survival analysis of HOX genes that predict (p < 0.05) patient survival. Curves reflect low (blue) and high (red) HOX gene expression. Y axis = Survival probability (0.0-1.0). X axis = Time (years 0-12). Bioinformatics data derived from The Human Protein Atlas (https://www.proteinatlas.org).

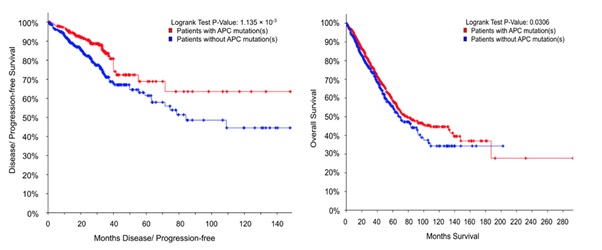 Figure 3: APC mutation predicts patient survival. Curves correspond to HOX gene methylation results in Table 1. PFS (left; n=361 vs 495) & OS survival (right; n=1567 vs 1005). APC mutant CRCs (red) & no APC mutation (blue).
Figure 3: APC mutation predicts patient survival. Curves correspond to HOX gene methylation results in Table 1. PFS (left; n=361 vs 495) & OS survival (right; n=1567 vs 1005). APC mutant CRCs (red) & no APC mutation (blue).
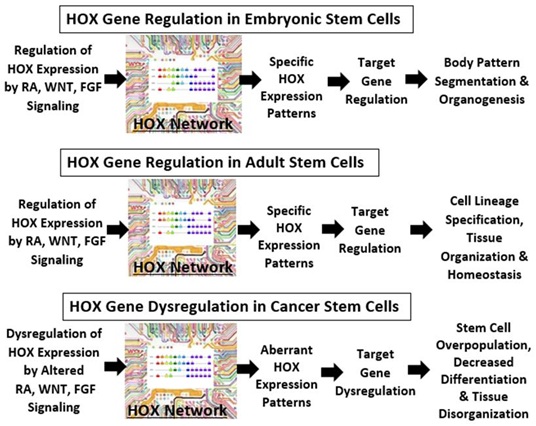 Figure 4: The schematic depicts how the HOX gene network might act like a computer motherboard “circuitry” in stem cells which processes and communicates information to other cellular processes that determine tissue phenotype. In embryogenic development, WNT, retinoic acid (RA), and Fibroblast Growth Factor (FGF) signaling regulates HOX gene expression (reviewed in [69]). The pattern of HOX gene expression, in turn, controls the expression of HOX target genes, which specifies body pattern segmentation and underlies organ development. In this view, the function of the HOX “circuitry” is variable and can depend on DNA methylation via genomic imprinting as well as spatial and temporal feedback mechanisms occurring during head-to-tail tissue formation in embryogenesis.
Figure 4: The schematic depicts how the HOX gene network might act like a computer motherboard “circuitry” in stem cells which processes and communicates information to other cellular processes that determine tissue phenotype. In embryogenic development, WNT, retinoic acid (RA), and Fibroblast Growth Factor (FGF) signaling regulates HOX gene expression (reviewed in [69]). The pattern of HOX gene expression, in turn, controls the expression of HOX target genes, which specifies body pattern segmentation and underlies organ development. In this view, the function of the HOX “circuitry” is variable and can depend on DNA methylation via genomic imprinting as well as spatial and temporal feedback mechanisms occurring during head-to-tail tissue formation in embryogenesis.
In adult tissues, the HOX gene network in stem cells continues to be controlled by WNT, RA, FGF, and other signaling pathways. However, the function of the HOX “circuitry” may differ in each of our various bodily tissues. In this way, the specific HOX gene expression patterns would differ from tissue to tissue. And, differences in HOX expression would lead to regulation of a different set of HOX target genes, which would control tissue phenotype by specifying SC differentiation into specialized cell lineages, maintaining cell organization and governing homeostasis.
In cancer tissues, an imbalance in RA, WNT, FGF, or other signaling pathways might lead to dysregulation of HOX gene expression in malignant stem cells. The function of the HOX “circuitry” may also become altered by HOX mutations, changes in DNA methylation and other epigenetic modifications. Such alterations would lead to aberrant HOX expression patterns and dysregulation of target gene expression. Ultimately, HOX gene dysregulation in cancer would lead to stem cell overpopulation, decreased cell differentiation, and disorganization of cells in tumor tissues.
MATERIALS AND METHODS
The bioinformatics analysis on overexpression, hyper-methylation, mutation, and copy number gain of HOX genes in CRCs was done through the COSMIC website (cancer.sanger.ac.uk/cosmic). Bioinformatics analysis to identify HOX genes that predict CRC patient survival was done through The Human Protein Atlas (https://www.proteinatlas.org). Our further analyses on HOX gene methylation were done as described in Supplemental Materials.
CONCLUSION
Our results indicate that the HOX gene transcriptional regulatory network, when dysregulated, plays a major role in the SC origin of CRC. Continued discovery of the mechanisms that explain how HOX genes regulate normal colon SCs and how dysregulation of HOX genes in cancer SCs drive CRC growth should provide insight into how new SC-targeted therapies might be designed for CRC. Thus, the significance of this research is that discovering how to modulate the function of the HOX gene circuitry may provide a mechanism to reverse changes in HOX gene expression patterns and to therapeutically target cancer stem cells.
ACKNOWLEDGEMENTS
We thank Dr. Nicholas Petrelli for his support at the Helen F. Graham Cancer Center and Research Institute, and Drs. Gilberto Schleiniger, Christopher Raymond, Lynn Opdenaker, Jeremy Fields as well as Katie Funk, Emily Russell, Victoria Oluwajuwon and Theresa Shafto for their valuable input and helpful discussions.
CONFLICT OF INTEREST
The authors do not have any conflicts of interest.
FINANCIAL SUPPORT
This study was supported in part by The Lisa Dean Moseley Foundation (BB), Cancer B*Ware Foundation (BB), and Cawley Center for Translational Cancer Research Fund (BO, CZ, BB, CF).
REFERENCES
- Bhatlekar S, Fields JZ, Boman BM (2018) Role of HOX Genes in Stem Cell Differentiation and Cancer. Stem Cells Int 2018: 3569493.
- Mallo M, Alonso CR (2013) The regulation of Hox gene expression during animal development. Development 140: 3951-3963.
- Smith J, Zyoud A, Allegrucci C (2019) A Case of Identity: HOX Genes in Normal and Cancer Stem Cells. Cancers (Basel) 11: 512.
- Chacón-Martínez CA, Koester J, Wickström SA (2018) Signaling in the stem cell niche: Regulating cell fate, function and plasticity. Development 145: 165399.
- Bhatlekar S, Viswanathan V, Fields JZ, Boman BM (2018) Overexpression of HOXA4 and HOXA9 genes promotes self-renewal and contributes to colon cancer stem cell overpopulation. J Cell Physiol 233: 727-735.
- Tharmapalan P, Mahendralingam M, Berman HK, Khokha R (2019) Mammary stem cells and progenitors: Targeting the roots of breast cancer for prevention. EMBO J 38: 100852.
- Takahashi K, Yamanaka S (2006) Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126: 663-676.
- Liu B, Wang T, Wang H, Zhang L, Xu F, et al. (2018) Oncoprotein HBXIP enhances HOXB13 acetylation and co-activates HOXB13 to confer tamoxifen resistance in breast cancer. Journal of Hematology & Oncology 11: 26.
- Seifert A, Werheid Df, Knapp SM, Tobiasch E (2015) Role of Hox genes in stem cell differentiation. World J Stem Cells 7: 583-595.
- Nolte C, De Kumar B, Krumlauf R (2019) Hox genes: Downstream "effectors" of retinoic acid signaling in vertebrate embryogenesis. Genesis 57: 23306.
- Carroll SB (1995) Homeotic genes and the evolution of arthropods and chordates. Nature 376: 479-485.
- Chen JD, Evans RM (1995) A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature 377: 454-457.
- Glass CK, Rosenfeld MG (2000) The coregulator exchange in transcriptional functions of nuclear receptors. Genes Dev 14: 121-141.
- Hörlein AJ, Näär AM, Heinzel T, Torchia J, Gloss B, et al. (1995) Ligand-independent repression by the thyroid hormone receptor mediated by a nuclear receptor co-repressor. Nature 377: 397-404.
- Lavinsky RM, Jepsen K, Heinzel T, Torchia J, Mullen TM, et al. (1998) Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proc Natl Acad Sci USA 95: 2920-2925.
- Mahony S, Mazzoni EO, McCuine S, Young RA, Wichterle H, et al. (2011) Ligand-dependent dynamics of retinoic acid receptor binding during early neurogenesis. Genome Biology 12.
- Kmita M, Duboule D (2003) Organizing axes in time and space; 25 years of colinear tinkering. Science 301: 331-333.
- Lewis EB (1978) A Gene Complex Controlling Segmentation in Drosophila. Genes, Development, and Cancer: 229-242.
- Nolte C, Jinks T, Wang X, Martinez Pastor MT, Krumlauf R (2013) Shadow enhancers flanking the HoxB cluster direct dynamic Hox expression in early heart and endoderm development. Dev Biol 383: 158-173.
- Gaertner B, Johnston J, Chen K, Wallaschek N, Paulson A, et al. (2012) Poised RNA polymerase II changes over developmental time and prepares genes for future expression. Cell Rep 2: 1670-1683.
- Lin C, Garrett AS, Kumar BD, Smith ER, Gogol M, et al. (2011) Dynamic transcriptional events in embryonic stem cells mediated by the super elongation complex (SEC). Genes Dev 25: 1486-1498.
- Begemann G, Schilling TF, Rauch GJ, Geisler R, Ingham PW (2001) The zebrafish neckless mutation reveals a requirement for raldh2 in mesodermal signals that pattern the hindbrain. Development 128: 3081-3094.
- Grandel H, Lun K, Rauch GJ, Rhinn M, Piotrowski T, et al. (2002) Retinoic acid signalling in the zebrafish embryo is necessary during pre-segmentation stages to pattern the anterior-posterior axis of the CNS and to induce a pectoral fin bud. Development 129: 2851-2865.
- Niederreither K, Subbarayan V, Dollé P, Chambon P (1999) Embryonic retinoic acid synthesis is essential for early mouse post-implantation development. Nat Genet 21: 444-448.
- Niederreither K, Vermot J, Schuhbaur B, Chambon P, Dollé P (2000) Retinoic acid synthesis and hindbrain patterning in the mouse embryo. Development, 127: 75-85.
- Deschamps J, Duboule D (2017) Embryonic timing, axial stem cells, chromatin dynamics, and the Hox clock. Genes & Development 31: 1406-1416.
- Deschamps J, van Nes J (2005) Developmental regulation of the Hox genes during axial morphogenesis in the mouse. Development 132: 2931-2942.
- Diez del Corral R, Olivera-Martinez I, Goriely A, Gale E, Maden M, et al. (2003) Opposing FGF and retinoid pathways control ventral neural pattern, neuronal differentiation, and segmentation during body axis extension. Neuron 40: 65-79.
- Diez del Corral R, Storey KG (2004) Opposing FGF and retinoid pathways: A signalling switch that controls differentiation and patterning onset in the extending vertebrate body axis. Bioessays 26: 857-869.
- Young T, Rowland JE, van de Ven C, Bialecka M, Novoa A, et al. (2009) Cdx and Hox genes differentially regulate posterior axial growth in mammalian embryos. Dev Cell 17: 516-526.
- Bel-Vialar S, Itasaki N, Krumlauf R (2020) Initiating Hox gene expression: in the early chick neural tube differential sensitivity to FGF and RA signaling subdivides the HoxB genes in two distinct groups. Development129: 5103-5115.
- Diez del Corral R, Breitkreuz DN, Storey KG (2002) Onset of neuronal differentiation is regulated by paraxial mesoderm and requires attenuation of FGF signaling. Development 129: 1681-1691.
- Neijts R, Amin S, van Rooijen C, Deschamps J (2017) Cdx is crucial for the timing mechanism driving colinear Hox activation and defines a trunk segment in the Hox cluster topology. Dev Biol422: 146-154.
- Hernandez RE, Putzke AP, Myers JP, Margaretha L, Moens CB (2007) Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development 134: 177-187.
- White RJ, Schilling TF (2008) How degrading: Cyp26s in hindbrain development. Dev Dyn 237: 2775-2790.
- Wilson V, Olivera-Martinez I, Storey KG (2009) Stem cells, signals and vertebrate body axis extension. Development 136: 1591-1604.
- Mallo M, Wellik DM, Deschamps J (2010) Hox genes and regional patterning of the vertebrate body plan. Dev Biol 344: 7-15.
- Bhatlekar S, Addya S, Salunek M, Orr CR, Surrey S (2014) Identification of a Developmental Gene Expression Signature, Including HOX Genes, for the Normal Human Colonic Crypt Stem Cell Niche: Overexpression of the Signature Parallels Stem Cell Overpopulation During Colon Tumorigenesis. Stem Cells Dev 23: 167-179.
- Bhatlekar S, Ertel A, Gonye GE, Fields JZ, Boman BM (2018) Gene expression signatures for HOXA4, HOXA9, and HOXD10 reveal alterations in transcriptional regulatory networks in colon cancer. J Cell Physiol 234: 13042-13056.
- Teo WW, Merino VF, Cho S, Korangath P, Liang X, et al. (2016) HOXA5 determines cell fate transition and impedes tumor initiation and progression in breast cancer through regulation of E-cadherin and CD24. Oncogene 35: 5539-5551.
- Raman V, Martensen SA, Reisman D, Evron E, Odenwald WF, et al. (2000) Compromised HOXA5 function can limit p53 expression in human breast tumours. Nature 405: 974-978.
- Ma H-M, Cui N, Zheng P-S (2020) HOXA5 inhibits the proliferation and neoplasia of cervical cancer cells via downregulating the activity of the Wnt/β-catenin pathway and transactivating TP53. Cell Death & Disease 11: 420.
- Depauw S, Lambert M, Jambon S, Paul A, Peixoto P, et al. (2019) Heterocyclic Diamidine DNA Ligands as HOXA9 Transcription Factor Inhibitors: Design, Molecular Evaluation, and Cellular Consequences in a HOXA9-Dependant Leukemia Cell Model. Journal of Medicinal Chemistry 62: 1306-1329.
- Lambert M, Alioui M, Jambon S, Depauw S, Seuningen IV, et al. (2019) Direct and Indirect Targeting of HOXA9 Transcription Factor in Acute Myeloid Leukemia. Cancers 11: 837.
- Palakurthy RK, Wajapeyee N, Santra MK, Gazin C, Lin, L, et al. (2009) Epigenetic Silencing of the RASSF1A Tumor Suppressor Gene through HOXB3-Mediated Induction of DNMT3B Expression. Molecular Cell 36: 219-230.
- Fu H, Fu L, Xie C, Zuo W-S, Liu Y-S, et al. (2017) miR-375 inhibits cancer stem cell phenotype and tamoxifen resistance by degrading HOXB3 in human ER-positive breast cancer. Oncol Rep 37: 1093-1099.
- Li X, Lin H, Jiang F, Lou Y, Ji L, et al. (2019) Knock-Down of HOXB8 Prohibits Proliferation and Migration of Colorectal Cancer Cells via Wnt/β-Catenin Signaling Pathway. Medical Science Monitor 25: 711-720.
- Wang T, Lin F, Sun X, Jiang L, Mao R, et al. (2019)HOXB8 enhances the proliferation and metastasis of colorectal cancer cells by promoting EMT via STAT3 activation. Cancer Cell International 19: 3.
- Shah N, Jin K, Cruz L-A, Park S, Sadik H, et al. (2013) HOXB13 Mediates Tamoxifen Resistance and Invasiveness in Human Breast Cancer by Suppressing ER and Inducing IL-6 Expression. Cancer Research 73: 5449-5458.
- Li B, Huang Q, Wei G-H (2019) The Role of HOX Transcription Factors in Cancer Predisposition and Progression. Cancers 11: 528.
- Li Y, Chao F, Huang B, Liu D, Kim J, et al. (2014) HOXC8 promotes breast tumorigenesis by transcriptionally facilitating cadherin-11 expression. Oncotarget 5: 2596-2607.
- Chao F, Zhang J, Zhang Y, Liu H, Yang C, et al. (2015) Embigin, regulated by HOXC8, plays a suppressive role in breast tumorigenesis. Oncotarget 6: 23496-23509.
- Shah M, Cardenas R, Wang B, Persson J, Mongan, et al. (2017) HOXC8 regulates self-renewal, differentiation and transformation of breast cancer stem cells. Molecular Cancer 16: 38.
- Bird A (2002) DNA methylation patterns and epigenetic memory. Genes & developmeID - 8711660, 16: 6-21.
- Fearon ER, Vogelstein B (1990) A genetic model for colorectal tumorigenesis. Cell 61: 759-767.
- Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, et al. (2006) CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature genetics 38: 787-793.
- Samowitz WS, AlbertsenH, Herrick J, Levin TR, Sweeney C, et al (2005) Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology 129: 837-845.
- Issa JP, Shen L, ToyotaM (2005) CIMP, at last. Gastroenterology 129: 1121-1124.
- Brotto DB, Siena ADD, de Barros II, da Costa e Silva Carvalho S, Muys BR, et al. (2020) Contributions of HOX genes to cancer hallmarks: Enrichment pathway analysis and review. Tumor Biology 42: 101042832091805.
- Paço A, de Bessa Garcia SA, Freitas R (2020) Methylation in HOX Clusters and Its Applications in Cancer Therapy. Cells 9: 1613.
- Bhatlekar S, Fields JZ, Boman BM (2014) HOX genes and their role in the development of human cancers. J Mol Med (Berl) 92: 811-823.
- Weidle UH, Birzele F, Kollmorgen G, Ruger R (2017) Long Non-coding RNAs and their Role in Metastasis. Cancer Genomics Proteomics 14: 143-160.
- Barnicle A, Seoighe C, Golden A, Greally JM, Egan LJ (2016) Differential DNA methylation patterns of homeobox genes in proximal and distal colon epithelial cells. Physiological genomics 48: 257-273.
- Iacopetta B (2002) Are there two sides to colorectal cancer? Int J Cancer 101: 403-408.
- Haenszel W, Correa P (1973) Cancer of the large intestine: Epidemiologic findings. Dis Colon Rectum 16: 371-377.
- Baran B, Ozupek NM, Tetik NY, Acar E, Bekcioglu O, et al. (2018) Difference Between Left-Sided and Right-Sided Colorectal Cancer: A Focused Review of Literature. Gastroenterology research 11: 264-273.
- Hanley MP, Hahn MA, Li AX, Wu X, Lin J, et al., (2017) Genome-wide DNA methylation profiling reveals cancer-associated changes within early colonic neoplasia. Oncogene 36: 5035-5044.
- Zhang X, Liu G, Ding L, Jiang T, Shao S, et al., (2018) HOXA3 promotes tumor growth of human colon cancer through activating EGFR/Ras/Raf/MEK/ERK signaling pathway. J Cell Biochem 119: 2864-2874.
- Qi L, Hu W, Zhou B (2019) HomeoboxC6 promotes metastasis of colorectal cancer by activating Wnt/b-catenin and inducing EMT. Basic gastroenterology 68: A44-A45.
- Yuan Y, Chen J, Wang J, Xu M, Zhang Y, et al., (2020) Development and Clinical validation of a Novel 4-gene prognostic Signature predicting survival in Colorectal Cancer. Frontiers in Oncology 10: 595.
- Zhang TM, Huang T, Wang RF (2018) Cross talk of chromosome instability, CpG island methylator phenotype and mismatch repair in colorectal cancer. Oncol Lett 16: 1736-1746.
- Lappin T, Grier D, Thompson A, Halliday H (2006) HOX genes: Seductive science, mysterious mechanisms. Ulster Med J 75: 23-31.
- Durston J, Zhu K (2015) A time space translation hypothesis for vertebrate axial patterning. Seminars in Cell & Developmental Biology 42: 86-93.
- Durston AJ (2019) Some Questions and Answers about the Role of Hox Temporal Collinearity in Vertebrate Axial Patterning. Front Cell Dev Biol 7: 257.
- Yu M, Zhan J, Zhang H (2020) HOX family transcription factors: Related signaling pathways and post-translational modifications in cancer. Cellular Signalling 66: 109469.
- Shah N, Sukumar S (2010) The Hox genes and their roles in oncogenesis. Nat Rev Cancer 10: 361-371.
- Luo Z, Rhie SK, Farnham PJ (2019) The Enigmatic HOX Genes: Can We Crack Their Code? Cancers (Basel) 11: 323.
- Geissler K, Zach O (2012) Pathways involved in Drosophila and human cancer development: The Notch, Hedgehog, Wingless, Runt, and Trithorax pathway. Ann Hematol 91: 645-669.
- Philippidou P, Dasen JS (2013) Hox Genes: Choreographers in Neural Development, Architects of Circuit Organization. Neuron 80: 12-34.
- Nordström U, Maier E, Jessell TM, Edlund T (2006) An Early Role for Wnt Signaling in Specifying Neural Patterns of Cdx and Hox Gene Expression and Motor Neuron Subtype Identity. PLoS Biol 4: e252.
- Kamkar F, Xaymardan M, Asli NS (2016) Hox-Mediated Spatial and Temporal Coding of Stem Cells in Homeostasis and Neoplasia. Stem Cells Dev 25: 1282-1289.
- Wheadon H, Ramsey JM, Dobbin E, Dickson GJ, Corrigan PM, et al., (2011) Differential Hox Expression in Murine Embryonic Stem Cell Models of Normal and Malignant Hematopoiesis. Stem Cells and Development 20: 1465-1476.
- Alharbi RA, Pettengell R, Pandha HS, Morgan R (2012) The role of HOX genes in normal hematopoiesis and acute leukemia. Leukemia 27: 1000-1008.
- Giampaolo A, Sterpetti P, Bulgarini D, Samoggia P, Pelosi E, et al., (1994) Key functional role and lineage-specific expression of selected HOXB genes in purified hematopoietic progenitor differentiation. Blood 84: 3637-3647.
- Kawagoe H, Humphries RK, Blair A, Sutherland HJ, Hogge DE (1999) Expression of HOX genes, HOX cofactors, and MLL in phenotypically and functionally defined subpopulations of leukemic and normal human hematopoietic cells. Leukemia 13: 687-698.
- Pineault N, Helgason CD, Lawrence HJ, Humphries RK (2002) Differential expression of Hox, Meis1, and Pbx1 genes in primitive cells throughout murine hematopoietic ontogeny. Experimental Hematology 30: 49-57.
- Ma I, Allan AL (2010) The Role of Human Aldehyde Dehydrogenase in Normal and Cancer Stem Cells. Stem Cell Rev Rep 7: 292-306.
- Modarai SR, Gupta A, Opdenaker LM, Kowash R, Masters G, et al., (2018) The anti-cancer effect of retinoic acid signaling in CRC occurs via decreased growth of ALDH colon cancer stem cells and increased differentiation of stem cells. Oncotarget 9: 34658-34669.
- Huang EH, Hynes MJ, Zhang T, Ginestier C, Dontu G, et al., (2009) Aldehyde Dehydrogenase 1 Is a Marker for Normal and Malignant Human Colonic Stem Cells (SC) and Tracks SC Overpopulation during Colon Tumorigenesis. Cancer Res 69: 3382-3389.
- Modarai SR, Opdenaker LM, Viswanathan V, Fields JZ, Boman BM (2016) Somatostatin signaling via SSTR1 contributes to the quiescence of colon cancer stem cells. BMC Cancer 16: 941.
- Nilsson HA (2001) The gut as the largest endocrine organ in the body. Ann Oncol 12: S63-S68.
- Schonhoff SE, Giel-Moloney M, Leiter AB (2004) Minireview: Development and Differentiation of Gut Endocrine Cells. Endocrinology 145: 2639-2644.
- Moran GW, Leslie FC, Levison SE, McLaughlin JT (2008) Review: Enteroendocrine cells: Neglected players in gastrointestinal disorders?. Therapeutic Advances in Gastroenterology 1: 51-60.
- Sternini C, Anselmi L, Rozengurt E (2008) Enteroendocrine cells: a site of `taste’ in gastrointestinal chemosensing. Curr Opin Endocrinol Diabetes Obes 15: 73-78.
- Shroyer NF, Kocoshis SA (2011) Anatomy and Physiology of the Small and Large Intestines. in Pediatric Gastrointestinal and Liver Disease. Elsevier 324-336.
SUPPLEMENTAL MATERIALS: HOX GENE AND DNA METHYLATION DATA ANALYSIS
Methods
DNA methylation data (beta value) generated via Illumina Infinium Human Methylation 27 (HM27) were downloaded from GDC Data Portal [1] and processed by our in-house Python script to generate the dataset for this analysis.
The methylation levels of all the HOX genes (totally 39 genes, with 78 protein-encoding transcripts) were explored. The correspondence of the gene information and protein-encoding Transcript IDs were extracted from ensembl.org [2].
To investigate the potential impact of APC frame-shift and/or stop-gained mutations on the global, and HOX genes, methylation patterns, comparisons were presented between CRC patients carrying APC frame shift and/or stop gained mutations (denoted as APC-mut group below), and CRC patients without APC frame shift or stop gained mutations (denoted APC-wt group below).
Results
Global methylation level distribution and the comparison between APC mutations
To have a global and comparative view of DNA methylation level under different APC mutation status, methylation beta value distributions are plotted in Figure S1. Particularly the whole genome methylation beta value distributions are compared between APC-mut group and APC-wt group in Figure S1.1. The same comparison of HOX genes is shown in Figure S1.2.
Figure S1: Methylation beta value distributions and their comparison between CRC patients with or without APC frame shift and/or stop gained mutations.
Figure S1.1: Methylation level distribution of all genes in the dataset, APC-mut group compared with APC-wt group;
Figure S1.2: Methylation level distribution of HOX genes, APC-mut group compared with APC-wt group.
It should be clear that the distributions of APC-mut group and APC-wt group have similar shapes and almost overlap with each other, in both Figure S1.1 and Figure S1.2. This indicates that the methylation patterns might not be vastly impacted by APC mutations. However, there are several noticeable differences (for example the far-left peak close to 0.0 in Figure S1.1, indicating that the APC-mut group might be slightly less methylated). Whether the similarity or difference pertains to HOX genes are further checked using two-sample t-tests below.
Two-Sample T-Tests on methylation beta values of HOX genes
To statistically evaluate the impact of APC mutations on the methylation levels of HOX genes, Two-sample t-tests were performed on their methylation beta values, between APC-mut group and APC-wt group.
To this end, a two-sample t-test was performed for the methylation beta values of each transcript, comparing between APC-mut and APC-wt groups. Corresponding t-statistics and p-values were determined (data available from corresponding author on request). Any p-value smaller than or equal to 0.05 was considered to be statistically significant.
Agreeing with the result of Figure S1, the t-statistic values indicating the difference of methylation levels between APC-wt and APC-mut groups revealed statistically significantdifferences for the following genes/transcripts.
- • The genes/transcripts that might have reduced methylation level upon APC frame-shift and/or stop-gained mutations include:
HOXA10, HOXA3, HOXA4, HOXB1, HOXB7, and HOXC8
- • The genes/transcripts that might have increased methylation level upon APC frame shift and/or stop gained mutations include:
HOXA2, HOXA5, HOXA6, HOXB8, and HOXD12
Patterns of hyper- and hypo- methylated genes
With the understanding that the methylation beta value distributions are generally similar between APC-wt and APC-mut groups, in this section we look into the general methylation patterns of the HOX genes, as which genes are more heavily methylated (hyper-methylated) and which genes are less methylated (hypo-methylated), regardless of APC mutation status.
Our analysis adopted a threshold for hyper-methylation (beta value ≥ 0.8) and a threshold for hypo-methylation (beta value ≤ 0.2), and categorized groups of transcripts sharing probe-detected CpG sites into the following categories (data available from corresponding author on request):
- • the genes/transcripts that consistently heavily methylated (or even hyper-methylated) including:
HOXA2, HOXB1, HOXB2, HOXD10, HOXD12, and HOXD4
- • the genes/transcripts that consistently less methylated (or even hypo-methylated) including:
HOXA1, HOXA11, HOXA7, HOXA9, HOXB13, HOXB4, HOXB7, HOXB8, HOXB9, HOXC12, HOXC4, HOXC5, HOXC8, HOXD1, HOXD11, HOXD13, HOXD8, and HOXD9
- • Some of the genes/transcripts have parts of their CpG sites consistently heavily/hyper- methylated and some other parts of CpG sites consistently less/hypo-methylated, including:
HOXA3, HOXA5, HOXB3, HOXC6, and HOXD3
Discussion
DNA methylation has been generally considered as a heritable modification across mitosis. Here with our finding that the genome methylation pattern is conserved across patients, and the assumption with the data collection process that each patient was sampled randomly (at different times/stages of the patient’s disease progress), we can conclude that DNA methylation patterns are likely to be heritable and conserved both within individual CRC patients and among CRC patient population, regardless of APC mutation status.
However this is not to say that the methylation pattern is the same between CRC patients and healthy individuals. Investigation into this direction would be a great extension to this analysis.
Although HOX genes are among the TCF targeted genes and (indirectly) influenced by APC [3,4], whether (and how) APC mutations might affect the methylation of HOX genes are still mechanistically unclear. With our data indicating that such an influence, though statistically exists, appears small (Figure S1), it might be of advantage if we can consider the methylation level to be a constant in a mathematical/statistical model involving the HOX genes methylation. On the other hand, it is also fair to admit that we do not know whether this statistically significant yet small difference we detected is biologically significant; to what degree should the patients’ clinical symptoms and prognosis be attributed to such a difference; or even how drastic a difference in methylation level can be considered biologically significant, in healthy or cancerous cells.
To shed a little more light into this direction, we cross-referenced the list of genes/transcripts that we have found to be statistically differentially methylated between APC-wt and APC-mut, with the list of genes that we have found to be either consistently heavily (hyper-) or less (hypo-) methylated. The rough result below shows how part of the biologically conserved methylation pattern changes upon APC high-impact mutations.
- • Genes/transcripts that are consistently heavily methylated and have the methylation level reduced upon APC frame shift and/or stop gained mutations include:
HOXB1
- • Genes/transcripts that are consistently heavily methylated and have the methylation level even increased upon APC frame shift and/or stop gained mutations include:
HOXA2 and HOXD12
- • Genes/transcripts that are consistently less methylated and have the methylation level even reduced upon APC frame shift and/or stop gained mutations include:
HOXC8
- • Genes/transcripts that are consistently less methylated and have the methylation level increased upon APC frame shift and/or stop gained mutations include:
HOXB8
Under the assumption that consistent methylation patterns are conserved and should reflect important functions on relevant genes expression level. The observed differences in methylation patterns of these genes corresponding to APC high-impact mutations are indicating that APC high-impact mutations might be affecting the organism/system through epigenetic mechanisms.
References
- The Cancer Genome Atlas Program, USA.
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, et al. (2020). Ensembl 2020. Nucleic Acids Research 48: D682-D688.
- Bienz M (2003) APC. Current Biology13: R215-R216.
- Hoier EF, Mohler WA, Kim SK, Hajnal A (2000) The Caenorhabditis elegans APC-related gene apr-1 is required for epithelial cell migration and Hox gene expression. Genes Dev 14: 874-886.
Citation: Osmond B, Zhang C, Dinh V, Boman BM, Facey COB (2020) HOX Genes and Cancer Stem Cells: Update on HOX expression, DNA Methylation, Biomarker Status, and Genetic Changes in Colorectal Cancer. J Stem Cell Res Dev Ther 6: 045.
Copyright: © 2020 Brian Osmond, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.