Primary Cutaneous Neuroendocrine Carcinoma: Case with an Atypical Location
*Corresponding Author(s):
Herrando Parreno GDepartment Of Radiation Oncology, Consorcio Hospitalario Provincial De Castellon, Castellon, Spain
Tel:+34 964.359700,
Email:gemas999@gmail.com
Abstract
Cutaneous primary Neuroendocrine Tumors (NETs) are rare carcinomas: only 10 cases have been described in the literature to date. They equally affect men and women, on average aged 60-90 years and there is a predilection for presentation in the head and trunk. NET presents cytomorphological and immunohistochemical characteristics typical of high-grade tumors with Neuroendocrine (NE) differentiation. Here we describe a clinical case of a primary coetaneous NET with an atypical presentation and details its therapeutic management.
CLINICAL CASE
We present the case of a 60-year-old male with a primary cutaneous NET located in the sacro-coxigenic region without evidence of any distant disease at time of diagnosis. The diagnostic confirmation was based on immunohistochemical determination of Neuron-Specific Enolase (NSE) and Chromogranin A (Cg A). We surgically respected the tumor and because the resection margins were close, we also administered adjuvant radiotherapy.
CONCLUSION
NETs are rare, slow-growing tumors which usually have a good prognosis. Surgery with curative intent is the gold standard treatment, and this may or may be associated with adjuvant radiotherapy when adverse factors are presents or when the tumor is considered unrespectable.
INTRODUCTION
Primary cutaneous Neuroendocrine Tumor (NETs) is a very rare [1,2] with an approximate global incidence of 5.25 in 100,000 cases [3]: only ten cases have previously been reported in the literature [4-13]. These neoformations can vary enormously, both in their biological and clinical behavior, in terms of their which location, degree of differentiation and secretory activity [14,15]. Despite this, most are low-grade neoplasms with an indolent course.
They are also called “carcinoid tumors” (well-differentiated neuroendocrine tumors) because they are characterized by the presence of neurosecretory granules which express specific markers, including Neuron-Specific Enolase (NSE), synaptophysin and Chromogranin A (Cg A) and CD56. In some cases, tumor cells secrete serotonin and other vasoactive substances which produce a carcinoid syndrome characterized by: abdominal pain, skin redness, diarrhea, sweating, palpitations etc [16].
NETs are derived from neural crest neuroendocrine cells called Kulchitsky cells and give rise to tumors with very different degrees of differentiation, varying from carcinoid tumors to the undifferentiated small cell (oat-cell) carcinomas. They are formed by epithelial cells which exhibit neuroendocrine differentiation and can present in any anatomical location, although they usually appear in the face and trunk. Their etiology is unknown and there is no known association with sun exposure, immunodepression or other predisposing factors.
The main diagnostic complexity in these cases lies in the distinction between primary cutaneous tumors and visceral neoplastic metastases because the latter may have a gastrointestinal or pulmonary origin but then cause cutaneous involvement. The mean age of presentation is between the sixth and ninth decade of life (mean 66.3 years) and studies have shown that the inicidence is similar between both sexes.
We recently treated a patient with a primary cutaneous neuroendocrine carcinoma, a different entity to Merkel-Cell Carcinoma (MCC) whose primary location was the sacro-coxygeal región. Metastases in the right lateral costal area and nasal pyramid also subsequently emerged. Given the infrequency of these tumor types and the atypical location and behavior of this instance, here we describe our clinical experience of the case.
CLINICAL CASE
A pelvic MRI performed in order to stage the tumor revealed alesión with a máximum diameter of 4cm with mesenchymal characteristics and calcium content, which affected the dermo-fatty layers of the sagittal and left parasagittal plane and presented cleavage planes with respect to muscular and bony structures (Figure 1).
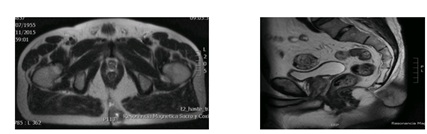
Figure 1: MRI of the transverse/sagittal grad/T2 planes, where the lesión described above was observed. The tumor had high calcium content and adhered to the dermo-fatty planes, precoxigeum, sagittal, left parasagittal, and cleavage planes with respect to muscular structures.
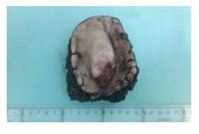
Given the presentation of a sacral área tumor with nonspecific characteristics, we then performed several differential diagnoses to distinguish between benign soft tissue lesions such as fibroids or malignant fibrosarcomas, chondromas, etc. A Fine-Needle Aspiration (FNA) biopsy of the lesion was positive for malignant tumor cells and so on 3 December, 2015; a block-excision of the lesion plus its margin was performed at the patients’s referral hospital. Macroscopically, the 7×4×2.5cm lesión was elevated well-delimited and had an unencapsulated polypoid surface with superficial ulceration. The lesion affected the dermis and subcutaneous cellular tissue and was located, 2mm from the deep margin, i.e., near the surgical margin (Figure 2).
Microscopically it was a diffuse trabecular carcinoma which affectedthe dermis and subcutaneous cellular tissue. The cells were large with nuclei containing fine chromatin threads and prominent nucleoli. The Immunohistochemical (IHC) study showed that the cells were positive for neuroendocrine markers (Cg A, synaptophysin and CD56). Cytokeratin 20 and TTF-1 staining was negative, thus a diagnosis of MCC or a pulmonary-origen metastases was excluded. The cell proliferation index (measured with Ki 67) was 80% (Figure 3 and 4).
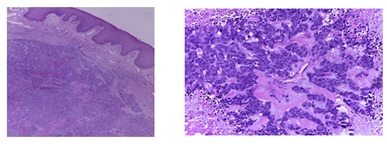
Figure 3: View of tumor microscopically: diffuse trabecular carcinoma cells were large with nuclei containing fine chromatin threads and prominent nucleoli.
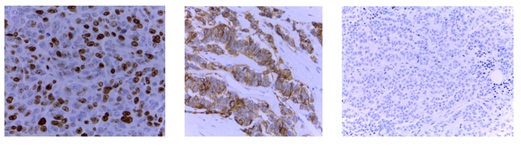
Figure 4: Evaluation of markers Immunohistochemical (IHC): Proliferation index Ki 67 CK20 negative NEmarkers CD56, positive.
There were no immediate complications in the postoperative period, except dehiscence of the surgical wound, which healed after a being restitched by plastic surgery service at the oncology hospital.
An extension study with PET/CT is requested without evidence of metabolically active tumor disease in other levels (Figure 5).
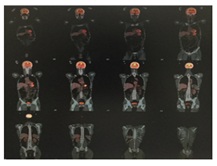
Figure 5: PET/CT performed with a Siemens Biograph-6-mobile True Point-True D-HD scanner with 7.5mCi of 18F-fludeoxyglucose (18F-FDG) according to our standard protocol.
We also requested blood analysis for tumor markers (carcinoembryonic antigen and carbohydrate antigen: CEA and Ca19.9) and neuroendocrine strain markers (Cg A and NSE) were in range at 6.04 and 8.5 respect. We did not determine the presence of 5-hydroxyindolacetic Acid (5-HIAA) in 24-hour urine even though others have shown than this, is useful for diagnosing and monitoring characinoid tumors [17].
More than three months after the initial surgical procedure, the wound had adequately healed and so we initiated adjuvant treatment with external radiotherapy using a Linear Particle Accelerator (LINAC) with prior to CT-simulation planning. The surgical bed in the sacal region plus the tumor margin (a 0.5cm bolus) were irradiated with 6Mv photons, using conventional fractionation until a total dose of 66Gy was reached (Figure 6). The patient presented acute grade-2 skin according to the Radiation Therapy Oncology Group (RTOG) scale [18].
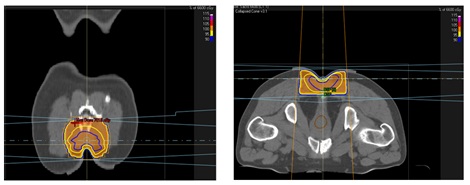
Figure 6: External Beam Radiotherapy (EBRT) treatment planimetry.
Three months later, at the end of the adjuvant treatment, the patient reported the presence of a new lesión. Further investigation revelaed it to be a 5×2cm bilobed subcutaneous lesion at the posterolateral right intercostal level, with non-specific ultrasound characteristics. Biochemical progression was observed, with elevation of neuroendocrine markers (NSE normal at 26.3ug/l and CrA was 2.70nmol/L); (IHC results were CKAE1AE3, synaptophysin and chromogranin positive and CK7, CK20 and TTF1 negative; Ki67 was 87%).
During the relapse diagnosis, on September 2, 2016, a nasal lesión appeared and was evaluated at the patient´s Ear, Nose and Throat (ENT) service at his local reference hospital. Examination showed it to be a 3x2cm subcutaneous, erythematous and violaceous tumor located on the nasal pyramid and infiltrating into the skin through to left nostril (Figure 7).
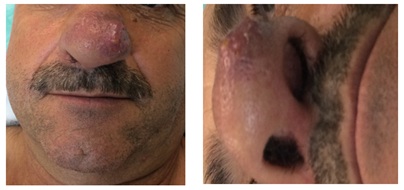
Figure 7: Initial nasal vestibular lesion.
Nasal endoscopy showed the presence of partially-collapsing, enlarged alveolar cartilage in the narrow part of the left nasal vestibule and fossa. A biopsy was taken, and the results were compatible with dermal infiltration by large cell neuroendocrine carcinoma cells with abundant mitosis and necrosis. In addition, the morphological and IHC results were matched the patient´s previous pre-biopsy tissue. Surgical treatment of the lesion was discarded as an option because of the progression of the disease.
Our extension study (performed before the emerged of this nasal lesión) included a pelvic MRI, which showed no evidence of local disease. Maxillofacial and cervical and thoracic CT showed the presence of a tumor with a 3.2cm diameter in the left nasal vetibule. There were no signs of any osteolysis or evidence of lymphadenopathy at the cervical and supraclavicular levels but multiple and bilateral lung metastases were observed (Figure 8).
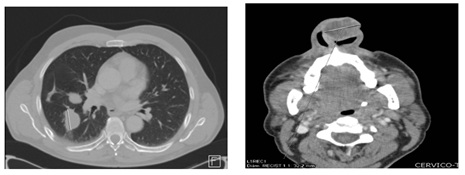
Figure 8: Cervic-thoracic CT scan of the área of the lesion described above.
Given the rapid and aggressive evolution of the disease and despite the lack of PET/CT results confirming systemic disease, we initiated a regieme of systemic chemotherapy with etoposide-platinum. The patient received two cycles, which he tolerated well without any associated toxicities.
The follow-up PET/CT performed after first cycle, showed the presence of a 4×3×3cm lesión in the left nasal ala (the maxium standardized uptake value (SUV max) was 10.54) a 3×5×4cm tumor in the rigth costal wall (the SUV max was 4.70), multiple foci of hypercaptation in the lung parenchyma ( the largest was in the lower right SUV max 8.74),bone metastatic lesions in the cervical spine, at the fourth left costal arch and the left coxal articulation (Figure 9). We did not perform an octreoscan scintigraphy because the patient was experiencing acute abdominal pain.
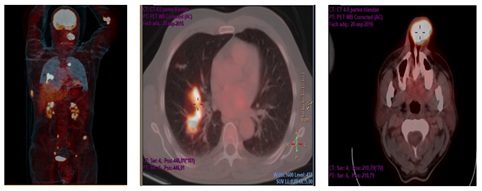
Figure 9: PET/CT performed with a Siemens Biograph-6-mobile True Point-True D-HD scanner with 7.5mCi of 18F-FDG according to our standard protocol.
Prior to the second cycle, there was a significant increase in both the lesión size: 7×5cm for the right costal tumor and 7×6cm for the left nasal tumor and of neuroendocrine markers expression in peripheral blood: NSE was 32.2ug (Figure 10).
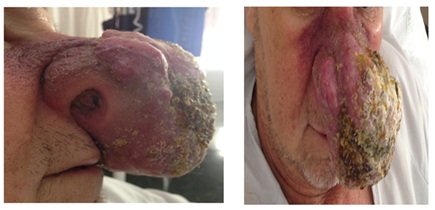
Figure 10: Current nasal excretory lesion.
Given the aggressive growth of the tumors, maxilofacial surgery was discarded because of the striking progression experienced during the systemic treatment. Therefore between October 17-28, 2016 we treated the left nasal and right costal Wall tumors (with palliative intention) with EBRT and prior CT-simulation, irradiating with 6Mv photons until we reached total dose og 30Gy (Figure 11).

Figure 11: EBRT planimetry image.
We also decided to initiate second-line chemotherapy with three cycles of a weekly regieme of placlitaxel, before any local or systemic progression appeared. However, the patient presented grade-4 toxicity (oral mucositis) with this scheme and required admission to Hospital for further treatment and support. In spite of this defensive prophylatic treatment, there was objective nasal progresion and so the previous chemotherapy regimen (weekly paclitaxel) was continued, until we obtained approval to initiate treatment with MSB0010718C (Avelumab).
DISCUSSION
Primary cutaneous NETs are rare with limited associated literature. Only 10 cases have been previously reported (Table 1 [19] and Table 2 [16]), many of which correspond to low-grade tumors (carcinoids), however, large cell carcinomas of this type are much rarer.
| Reference | Age (Years) and Gender | Location | Tumor Size (cm) and Duration (Years) | Clinical Diagnosis | Treatment | Follow-Up (Years) | Recurrence |
| Van Dijk et al. [4] | 72, female | Scalp | 3/10 | Pilar cyst | Excision | 3.5 | No |
| Smith and Chappell [5] | 62, female | Anterior chest | 1.5/10 | NC | Excision | 0.9 | No |
| Colllna et al. [6] | 80, male | Scalp | 4/1 | NC | Excision | 1.75 | No |
| Bart et al. [7] | 40, male | Epigastrium | 1/4 | Neurofibroma | Excision | 1.9 | No |
| Sakamoto et al. [8] | 87, female | Anterior chest | 9.5/60 | NC | Excision | NC | No |
| Courvllle et al. [9] | 60, male | Anterior chest | NC/NC | NC | Excision | 4 | No |
| MacKenzie et al. [10] | 70, female | Scalp | 3/NC | Sebaceous cyst | Excision | 6 | Yesa |
| Cokonls et al. [11] | 64, male | Middle back | 2/2 | NC | Excision | 1 | No |
| Eloy-Garcia Carrasco et al. [12] | 58, female | Scalp | 4/1 | NC | Excision | 1 | No |
| Kropinak et al. [13] | 70, female | Eyelid | 1/3 | NC | Excision | 0.67 | No |
aRegional lymph node metastasis was present at the time of the initial diagnosis, 4 and 6 years thereafter.
| Reference | Age MfF | Clinical | Ultra structure | Treatment | Course |
| Van Dijk C et al. [4] | 72 yr female | Cystic lesion on forehead with flushing | Argophyllic granules | Exertion | Rushrfig resolved. No recurrence * 1 yr |
| Sakamoto F et al. [8] | 75 yr Female | Hyperpigmented nodule with periumbilical induration. Elevated 5HIAA. Associated hyperparathyroidism | Strongly positive argentaffin reaction | Exertion | Not given in report |
| Hoefnagel CA [20] | 80 yr male | Nodule behind right ear * 12m | Argophy Ik granules.Chromgranin positive Negative argentaffin reaction | Wide exocosion | No recurrence 21 months later |
| Bart RS et al. [7] | 40 yr male | Red-tan soft nodule on abdomen * 4 years | Argophylk large and varied granules. NSE. keratin +ve | Wide exocosion | Alive and well 2 years later |
| Smith PA et al. [5] | 62 yr female | Firm Nue-rth nodule on chest | Argophjrfcc granules. Postwe argentaffin reaction | Wide exocosion | Alive at 4 years. Well apart from claudication |
| Courville P et al. [9] | 60 yr male | Erythematous nodule on chest | Argophyfcc granules. Positive argentaffin reaction.Chromogranin A. NSE | Excision | Alive without recurrence 4 years after excision |
| Mackenzie DN et al. [10] | 70 yr female | Hard node on scalp with metastasis to occipital lymph node | Syruptophysin. NSE. keratin.CEA +ve | Excision of lesion and ocopital lymph node | 4 years later further lymph node metastasis.Metastatic noderecurrence 2 years later |
Table 2: Primary cutaneous neuroendocrine tumours [16].
NETs present as single cutaneous nodules, which adhere to the skin and have a yellowish appearance due to their high lipid content. They are usually, dome-shaped or protrude above the skin surface and extending into the Subcutaneous Tissue (SCC), in most cases. They express one or more neuroendocrine markers which can include cytokeratins AE1/AE3, CK7, MNF and CD56. Carcinoid tumors develop and grow slowly: most lesions are asymptomatic, although some have episodes of self-limiting bleeding, tenderness or pruritus. They may be accompanied by clinical symptoms of carcinoid syndrome which is secondary to the release of 5-hydroxytryptamine (5HT). They have a favorable prognosis because the percentage of recurrences is low and they rarely metastasize to distant sites [19].
Large cell neuroendocrine carcinomas NETs are even rarer and are harder to recognize and so their incidence is probably underestimated. These tumor cells are large and polygonal and have a high nucleo-to-cytoplasmic ratio, nuclei containing fine chromatin threads and a prominent nucleolus, but lack the characteristic paranuclear CK20 staining of MCCs. They have an aggressive phenotype similar to analogous lung cancer but because they are rare their prognosis is not known. Likewise, their optimal specific therapy is also unknown, although some cases have treated with radiotherapy.
The diagnosis is confirmation via histopathological and IHC techniques, however, imaging techniques such as CT and MRI, are also required to rule out systemic involvement. This consideration is key, because the presence of distant disease is a crucial prognostic factor and is associated with at least a 20% rediction in the predicted 5-year survival time (from 70% to less than 50%) [2]. Diagnosis can be complex for small tumors, when usngconventional imaging techniques and so Metaiosobenzylguanidine (MIGB) scintigraphy scans using somatostatin-octreotide analogues are also used. MIGB scans have a sensitivity of around 72-87% but a lower specificity [21,22].
In addition to these aforementioned difficulties, the diagnosis of primary neuroendocrine cutaneous tumors is complicated by the rarity of this type of neoplasia and the histopathological complexities associated with it. Therefore they are diagnosed by exclusion, first discarding other possible tumoral origins. The primary differential diafnosis is made against MCCa highly malignanti small cell tumor which expresses CK20 in is t cytoplasm and which contains cutaneous neuroendocrine tumor metastases from other locations [19].
Coetaneous metastases produced by visceral neuroendocrine tumors are having a low incidence; however, they should be differentiated from primary cutaneous NETs. The formed rare multifocal lesions with a limited 1-2cm size and also present clinical symptoms derived from the primary visceral tumor (e.g., digestive or respiratory problems) [23-25]. Radiological test are required to rule out the presence of primary tumors originating in, another organ and Thyroid Transcription Factor 1 (TTF1) staining is used to rule out a pulmonary origin. In our case, TTF1 was negative and no visceral neuroendocrine neoplasia was clinically or radiologically evident.
Primary neuroendocrine coetaneous tumors, which have an incidence accounting for around 1% of malignant skin tumors, (although this incidence has increased probably because of increased population aging and greater sun exposure) [20] should also the differentiated from MCCs. MCC is more common in men, in which the median age at diagnosis is 76 years. Histological, it is characterized by diffuse tumor growth with a trabecular pattern, the presence of small uniform cells, with little cytoplasm surrounding an ovoid nucleus with fine threads of dispersed chromatin, the absence of a nucleolus and a high mitotic duplication rate: it shows a characteristic pronuclear staining pattern when stained with CK20.
MCC is also characterized by its aggressive behavior with local recurrences, frequent distant metastases and a poor response to conventional therapies [26,27]. The 5-year survival rate is around 50% for patients with NETs with nodal or distant involvement. The mean survival rate decreased to only 5 months for patients with MCCs.
The initial therapeutic approach, for patients with NETs is surgical resection which represents a potentially curative treatment. According to the recommendations, adjuvant radiotherapy should also be administered where nearby marins are affected or in the case of unresectable tumors.
The case we describe here had been developing over 20 years, with an exponential increase in tumor size observed in the last 5 months, suggesting that this low-grade NET had undergone a transformation to a high-grade carcinoma. Histological, this hypothesis is supported by the presence of a well-delimited 2cm area of intense fibrosis, calcification and bone metaphase containing fewer tumor cells than would normally correspond to the primary tumor site. Another possibility is that this was a metastatic tumor, what was undetectable by the available diagnostic modalities, similar to the case described in Jedrych J et al. [19].
In order to reach a diagnosis of visceral neoplasia, it may be useful to perform a MIGB scan using a somatostatin-octreotide analogue, as described above. In our case, we could not perform this test because the patient presented intense abdominal pain that required admission to hospital. Therefore we instead performed a PET/CT study.
We treated the patient with block surgical resection of the macroscopic lesion and, completed the treatment with external radiotherapy, thus minimizing the possibility of residual disease remaining in the surgical bed. Our patient then received standard systemic treatment without presenting any significant acute toxicity. This treatment consisted of a regieme of etoposide-cisplatin chemotherapy, which is applied in patients with poorly-differentiated unresectable high-grade (G3) neuroendocrine carcinomas or in those with systemic disease. The response rate is 40-80% with median response duration of 8-11 months and a median survival of 9-15 months [28]. Other potential therapeutic agents were doxorubicin, sunitinib or everolimus [29]. Before progression of the systemic disease, we started weekly second- line, paclitaxel treatment, although thos showed no clinical benefit and produced sever grade 4 mucosal toxicity (oral mucositis).
Given the rapid progression of the disease and absence of any benefits derived from abovementioned second-line treatment, we proposed a new line of treatment with Avelumab. This anti-PD-L1 monoclonal antibody has a suggested application in stage-4 MCCs which have progressed following cytotoxic chemotherapy, as reflected in its recently published phase 2 clinical trials [30]. Its use is also valid for solid metastases or locally-advanced gastric tumors, non-small cell lung tumors, ovarian tumors [31]. However we started administering a second round of the standard chemotherapy regieme described above, until our local Health Authority approved the use of this immunological therapy for our case.
CONCLUSION
We consider this case to be of relevance, not only because of its histology but also beacuses of its atypical location and its torpid development over 20-year. The lesión had significantly grown in size in the months leading up to the patient´s presentation and it had transformed into a large cell NET with a proliferation rate of 80%.
One possible explanation for this evolution is that it was initially a low-grade atypical NET carcinoma that transformed into high-grade NETs. This hypothesis ties in well with the histology data showing the presence of a well-delimited area of intense fibrosis, calcification and bone metaphase with less temporal cellularity than would normally correspond to the area of the primary tumor.
When it was diagnosed as an aggressive tumor (because of its high proliferation index, close margins, coetaneous involvement and diffuse positivity for NE markers) we chose to treat the patient with surgery associated with adjuvant treatment and EBRT in order to gain better local control. But when systemic progression and grade-4 toxicity presents after application of these two treatment lines we decided to request immunological therapy with Avelumab.
REFERENCES
- Kulke MH, Mayer RJ (1999) Carcinoid tumors. New Eng J Med 340: 858-868.
- Modlin IM, Sandor A (1997) An analysis of 8305 cases of carcinoid tumours. Cancer 79: 813-829.
- Yao JC, Hassan M, Phan A, Dagohoy C, Leary C et al. (2008) One hundred years after “carcinoid”: epidemiology of and prognostic factors for neuroendocrine tumors in 35,825 cases in the United States. J Clin Oncol 26: 3063-3072.
- Van Dijk C, Ten Seldam RE (1975) A posible primary cutaneous carcinoid. Cancer 36: 1016-1020.
- Smith PA, Chappell RH (1985) Another posible primary carcinoid tumour of skin? Virchows Arch A Pathol Anat Histopathol 408: 99-103.
- Collina G, Quarto F, Eusebi V (1998) Trabecular carcinoid of the skin with cellular stroma. Am J Dermatopathol 10: 430-435.
- Bart RS, Kamino H, Waisman J, Lindner A, Colen S (1990) Carcinoid tumor of skin:report of a posible primary case. J Am Acad Dermatol 22: 366-370.
- Sakamoto F, Ito M, Matumura G, Sato Y, Kimura S (1991) Ultraestructural study of a mucinous carcinoid of the skin. J Cutan Pathol 18: 128-133.
- Courville P, Joly P, Thomine E, Ziade J, Soubrane JC (2000) Primary cutaneous carcinoid tumour. Histopathology 36: 566-567.
- Mackenzie DN, McCormick CS, Morris RJ (2003) Lymph node metástasis from a primary skin carcinoid tumour. Br J Plast Surg 56: 718-721.
- Cokonis CD, Green JJ, Manders SM (2004) Primary carcinoid tumor of the skin. J Am Acad Dermatol 51: 146-S148.
- Eloy-Garcia Carrasco C, Benguigui Benadiva J, Martinez Garcia S, Sanz Trelles A, Palacios S (2006) Atypical primary carcinnoid tumour of the skin. J Cutan Pathol 33: 32-34.
- Kropinak M, Sims L, Iacob C, McCormick SA, Milman T (2009) Primary typical carcinoid tumor of the eyelid. Ophthal Plast Reconstr Surg 25: 318-320.
- Akerström G, Hellman P (2007) Surgery on neuroendocrine tumours. Best Pract Res Clin Endocrinol Metab 21: 87-109.
- Tomulescu V, St?nciulea O, Dima S, Herlea V, Stoica Mustafa E et al. (2011) [Diagnosis and surgical management in gastrointestinal neuroendocrine tumors]. Chirurgia 106: 151-161.
- Hyer SL, McAleese J, Harmer CL (2007) Neuroendocrine carcinoma arising in soft tissue: three case reports and literatura review. World J Surg oncol 5: 77.
- SEQC, Lab Tests Online. American Association for Clinical Chemestry. SEQC.
- RTOG Foundation Inc, RTOG/EORTC Late Radiation Morbidity Scoring Schema. RTOG Foundation Inc, Philadelphia, USA.
- Jedrych J, Pulitzer M (2014) Primary carcinoid tumor of the skin: a literature review. Int J Surg Pathol 22: 129-135.
- Hoefnagel CA (1995) MIGB and radiolabeled octreotide in neuroendocrine tumors: Q J Nucl Med 39: 137-139.
- Kaltsas G, Korbonits M, Heintz E, Mukherjee JJ, Jenkins PJ et al. (2001) Comparison of somatostatin analog and meta-iodobenzylguanidine radionuclides in the diagnosis and localization of advanced neuroendocrine tumors. J Clin Endocrinol Metab 86: 895-902.
- Keane J, Fretzin DF, Jao W, Shapiro CM (1980) Bronchial carcinoid metastatic to skin. Light and electron microscopic findings. J Cutan Pathol 7: 43-49.
- Rodriguez G, Villamizar R (1992) Carcinoid tumor with skin metástasis. Am J Dermatopathol 14: 263-269.
- Caplin ME, Buscombe JR, Hilson AJ, Jones AL, Watkinson AF, et al. (1998) Carcinoid tumour. Lancet 352: 799-805.
- Hussain SK, Sundquist J, Hemminki K (2010) Incidence trends of squamous cell and rare skin cancers in the Swedish national cancer registry point to calendar year and age-depent increases. J Invest Dermatol 130: 1323-1328.
- Sibley RK, Dehner LP, Rosai J (1985) Primary neuroendocrine (Merkel cell) carcinoma of the skin. A clinicopathologic and ultrastructural study of 43 cases. Am J Surg Pathol 9: 95-108.
- Perlman JP, Lumadue JA, Hawkins AL, Cohen K, Colombani P, et al. (1995) Primary cutaneous neuroendocrine tumors. Diagnostic use of cytogenetic and MIC2 analysis. Cancer Genet Cytogenet 82: 30-34.
- GETNE: Grupo Español de Tumores Neuroendocrinos. Barcelona, Spain.
- Kuehr T, Thaler J (2016) Current protocols and targeted therapies.Chemotherapy Protocols.
- Kaufman HL, Russell J, Hamid O, Bhatia S, Terheyden P et al (2016) Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 tria. Lancet Oncol 17: 1374-1385.
- Pfizer Inc (2016) ASCO 2016: Pivotal Avelumab Study Shows Positive Results in Metastatic Merkel Cell Carcinoma. Pfizer Inc, New York, USA.
Citation: Parreno GH, Macías VM, Santander RL, Ferrero JLE, Caballer JB et al. (2017) Primary Cutaneous Neuroendocrine Carcinoma: Case with an Atypical Location. J Cancer Biol Treat 4: 011.
Copyright: © 2017 Herrando Parreno G, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.